Немного молекулярной биологии, post 1
Дисклеймер: в рамках РТД обсуждался вопрос об полезных основах современной биологии для ТГ, в силу своей специальности я начну кратко описывать в адаптированной неформальной форме основные (и примечательные для ТГ) моменты. Биоинженер, помни! Ты должен всё это знать и ты мне это расскажешь на коллоквиуме, зачёте и экзамене!
Нет нужды напоминать основы биохимии, все помнят что основными компонентами живого являются белки, жиры, нуклеиновые кислоты, вода, неорганические вещества и продукты обмена веществ (метаболизма) как всяческие производные вышеназванного; при этом белки это функциональные исполнители всего-всего, жиры - главный компонент мембран и запас энергии, углеводы используются как носитель/оперативный запас энергии и для конструкционных материалов, нуклеиновые кислоты главным образом являются информационным и регуляторым агентом, вода это основная среда для жизни и неорганические вещества обеспечивают физико-химический "фон" чтобы это всё работало и ещё являются специфическими компонентами для многих ферментов.
Также все помнят основы молекулярной биологии, что ДНК это главное (и единственное) хранилище информации в клетке (хромосомы), изредка используемое как агент переноса генов (плазмиды и прочие векторы); РНК используется как временный носитель информации (мРНК), реже как регулятор чего-нибудь (РНК-интерференция, рибопереключатели) и ещё реже как исполняющий агент (рибозимы) за одним громадным исключением (рибосома); что белки являются главенствующими компонентами живого, которые что-то делают, поскольку делаю они всё - превращение метаболитов, перенос веществ (через мембраны и пр.), детекция веществ, передача сигнала, детекция физико-химических условий, механическую работу, регуляция других компонентов,...
В этом посте мы, после того как вспомнили основной бэкграунд, рассмотрим "центральную догму молекулярной биологии" вообще и что там насчёт контроля, который необходим для управления биологическими процессами и системами.
Это пафосное название, описывающее поток генетической информации, имеющий место (хотя-бы частично) в любом земном живом (или не очень, если речь о вирусах) объекте. Вот картинка:
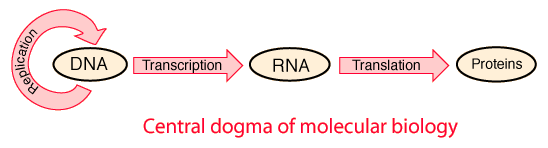
Закодированная в ДНК информация (геном, вся хромосомная ДНК, геном митохондрий и хлоропластов рассматривают отдельно, плазмиды и торчащие в клетке ДНК-овые вирусы это не_геном) переводится (считывается, транскрибируется) с помощью транскрипции в РНК (транскриптом, всё что транскрибируется с ДНК), если РНК кодирующая (матричная РНК, в ней закодирована последовательность белка, аминокислотная последовательность - КО), то по ней синтезируется (мРНК транслируется) белок (протеом, совокупность всех белков). Схема может быть продолжена, хотя в классическую догму это обычно не включают: совокупность метаболических (упрощая - химических) процессов, производимых в подавляющем большинстве белками, образует метаболом (все метаболические процессы и вещества, в них участвующие), который и реализует фенотип организма (существует редко применямый термин "фенОм", но это затрагивать мы не будем). Вопросы репликации пока поднимать не будем и перейдём к более подробному рассмотрению каждого шага.
Транскрипция
Процесс синтеза РНК по цепи ДНК с помощью РНК-полимеразы (фермента, строящего цепочку из чего-нибудь, здесь из РНК), синтезируются РНК относительно (в смысле, хромосом) небольшой длины на уровне тысяч нт (нуклеотидов), изредка достигая сотен тысяч нт и более. У не_бактерий каждый (нормальный) ген транскрибируется в одной РНК, много генов в одном фрагменте РНК бывает только у бактерий (и вирусов). Модульность "один ген - одна его РНК" (у бактерий часто "одна серия генов - одна их РНК") обеспечивается за счёт наличия более-менее чётких управляющих элементов, техническая аналогия - РНК это ксерокопия чертежа со страницы большой книги. Картина полного цикла транскрипции формального гена:

Транскрипция начинается (инициируется) со специализированных относительно чётких мест в геноме, стоящих недалеко перед геном, они называются "transcription start site" - TSS (да, КО был здесь), транскрипция прекращается (терминируется) на специальных местах остановки, именуемых терминаторами, хотя с ними чёткости меньше. Специальные места в геноме, где располагаются TSS, являются более-менее определёнными последовательностями нуклеотидов - промотерами, точнее промотерными областями генов, с которыми связываются белки, помогающие полимеразам начинать транскрипцию - факторы инициации транскрипции. В процессе исполнения транскрипции (элонгации) нет почти ничего интересного, это опустим. Терминация заканчивается "где-нибудь, главное за геном", потому что этого, чаще всего, достаточно для нормальной работы гена. Важно что с одного куска ДНК (а их всего 1 или 2 на весь геном, исключая повторяющиеся элементы) можно сделать много транскриптов, что обеспечивает "усиление сигнала". Сплайсинг, как фишка только эукариот (об археях не говорим), будет чуть позже.
Трансляция
Процесс синтеза белка по мРНК выполняется с помощью рибосомы (рибозима со вспомогательными белками), использующей тРНК (специальные небольшие РНК с присоединёнными аминокислотами и антикодонами), которая пробегает по мРНК, длины аминокислотных цепей обычно на уровне сотен звеньев. Трансляция выполняется как перевод трёхбуквенных сочетаний нт (кодонов) в аминокислотную последовательность, соответствия тем, что и называется "генетический код". Картинки не будет.
Трансляция инициируется со специального сигнального кодона (стартовго кодона, в общем говоря их несколько, но главный A-U-G), начинать именно с нужного (случайных сочетаний AUG всегда немало) стартового кодона (с которого начинается кодирующая часть РНК) рибосоме помогает контекст (последовательность) и белки (факторы инициации), для разных организмов механизмы отличаются. Цикл элонгации трансляции трогать не будем. Терминация трансляции выполняется за счёт специальных белков на сигнальном кодоне остановки (стоп-кодоне). Опять-же, стоит отметить что по одной мРНК могут пробежать много рибосом, так можно получить много белка с одной молекулы, что обеспечивает "усиление сигнала" и на этапе трансляции.
Последующие процессы уже не имеют обобщённых схем, потому говорить "в общем" уже бессмысленно, перейдём к методам исследований на каждом уровне:
На уровне ДНК - секвенирование (прочтение) геномной ДНК (DNA-seq), уже давно ставшее рутинным (хотя всё ещё дорогостоящим) методом, позволяет получать геном практически полностью для любого объекта живой природы (за исключением некоторых растений), но важно понимать что "прочтение" не значит "понимание" (геномика), одним секвенированием на все вопросы не ответить, потому что нужна информация о том, что именно из себя представляют и как работают все сигнальные и смысловые элементы в последовательности ДНК, а это устанавливается только экспериментами на следующих уровнях. На ранних этапах (первые маленькие геномы в 80-90х годах и большие к 00м) прочтение геномов давало много новых знаний и было "высоконаучно", сейчас (точнее, уже примерно лет 10) это не является передовыми научными исследованиями. Для новых организмов это технический конвейер получения данных (для краткости NGS) для эволюцинистов (и какие-то частности для многих остальных), а для человека это генетические медицинские исследования. В последнем случае это тоже конвейер получения данных для медицинских генетиков, давно являющися рутиной и практически никаких новых открытий не дающий, кроме роста баз сопоставлений генетических вариаций и патологий. Стоит отметить шквалы работ среднего качества по полногеномным исследованиям, в которых оценка влияния обнаруженных геномных вариаций (найденных с помощью методологии "перелопатим всё, найдём что-нибудь") при какой-нибудь патологии не имеет ничего, кроме небольших статистических корреляций, без какой-либо вообще проверки на биологическую адекватность, разбирательства в механизме и проверки на моделях, из-за чего бОльшая часть генетических вариантов в различных базах данных имеет либо никакой эффект (что, обычно, правда), либо слабый эффект, достоверность которого писана вилами на воде. Таким образом, основная сложность "прочитать" вполне преодолена, но она не помогает "понимать". Также это нужно помнить тем, кто говорит "расшифровка генома" (хотя это чисто биоинформатическая работа) заместо "секвенирования генома". Замечание о том, что и сейчас полностью целиком прочитать и собрать геном, например, человека всё ещё не возможно будет описано в следующих постах, а про такое старьё как SNP-чипы я не буду тут рассказывать.
На уровне РНК - измерение количества РНК (для количественной оценки транскрипции как этапа экспресси гена), выполняемое или количественным ПЦР (давно ставшим уже клиническим), если речь по несколько конкретных РНК, либо экспрессионными чипами, если нужно измерять сразу очень много разных известных РНК из известных объектов (ограниченный и устаревающий метод), либо с помощью секвенирования РНК (RNA-seq), которое и прочитает всё, и измерит сколько чего. Собственно в исследованиях на уровне систем клетки хорошо именно последнее, хотя метод дороже чем DNA-seq (который и так дорог), основное его применение - смотреть на то, как меняется транскриптом клетки при каких-либо процессах (воздействия лекарства, вируса, повреждения, дифференциация), т.е. какие гены начали транскрибироваться больше/меньше и ещё что-нибудь, в роде альтернативных сплайс-форм. Позволяет изучать сигнальные каскады, клеточные механизмы и много других важных вещей. Метод тоже хайповый (как и любой ЧегоНибудь-seq), потому есть очень много работ (особенно онкологических, столько, что напрягает читать заголовки журналов в подписках) где любят секвенировать, машинно строить гипотезы о взаимосвязях генов xyz2 и qwe5 при воздействии/патологии H8 на основании статистической корреляции между изменениями экспрессии xyz2 и qwe5 без какого-либо дополнительного разбирательства в механизме. О разных видах RNA-seq и напрягах исследователей будет в специальном после про секвенирование.
Комбинированные методы:
Просто прочтение ДНК и РНК даёт информацию о последовательностях и о их количестве, но это не всё, что имеет место с нуклеиновыми кислотами. С ДНК связываются белки, такие как полимеразы, факторы транскрипции, подавители транскрипции, компоненты систем репарации повреждений (о них будет отдельный пост), белки для репликации, для паковки ДНК (гистоны в случае эукариот), механических манипуляций... и с РНК тоже много чего может связываться (даже больше, РНК гибче), причём связываются эти белки, как правило, с некоторой специфичностью по последовательности (что чаще всего) или структурному мотиву (вспоминаем про комплементарность). Важно знать каков этот мотив последовательности, поскольку это позволяет понимать и предсказывать пути генетической регуляции. Для таких задач применяются методы иммунопреципитации хроматина и РНК (ChIP и RIP), когда ДНК/РНК вместе с белками выделяется, дробится и с помощью антител к интересующему белку вытаскиваются куски ДНК, связавшиеся с нашим белком. Секвенирование этих фрагментов (ChIP-seq и RIP-seq) даёт нам селективное обогащение по кускам ДНК, с которыми связался интересующий белок, так и выясняется где в геноме он садится и каков мотив связывания белка. Для факторов транскрипции, например, это даёт предположения о том, какие гены он может включать.
Вспоминая очень модный нынче тренд "эпигенетика" можно вспомнить не только про ChIP-seq к гистонам (белки, на которые намотана запакованная ДНК в хромосомах) разного вида, но и про химические модификации ДНК (видов которых очень мало, если мы не поливаем клетки ипритом и этиленоксидом), точнее про метилирование. Грубо говоря, метилирование ДНК у эукариот позволяет перманентно запаковать и подавить транскрипцию, перекосы с общим и специфическим метилированим имеют место во многих серьёзных проблемах, таких как старение и рак (потому и хайпа много). С помощью химической модификации (бисульфит обычно, называют BS-seq) можно сделать различимыми при секвенировании метилированные и нет нуклеотиды (технически обработка данных метода значительно труднее и RNA-seq и ChIP-seq, как пришлось как-то убедиться самому). И ещё много инетересных методов, основанных на NGS.
На уровне белка - а вот тут всё плохо, поскольку высокопроизводительного (за счёт массивного параллелизма, как в NGS) метода для секвенирования белков не существует (потому что нет процесса копирования полипептидной цепи, а все работающие секвенаторы сейчас используют именно этот процесс для ДНК). Старые методы разделения белков не справляются с очень многими тысячами белков полной протеомы кого угодно (и в случае высшых эукариот это многообразие осложняется альтернативными формами мРНК), старые методы секвенирования требуют большого количества белка (а неизвестный белок нельзя просто взять и накопировать, в отличии от ДНК и РНК), единственным исключением является метод 2D-GE (за попытку использования этого метода кое-кто может попытаться утопить вас в форезной ванне), которй можно хотя-бы пытаться применять к протеому кого-нибудь, да и то, он сопряжён с масс-спектрометрией... ...на основе которой многие (с некоторым успехом) реализуют метод, всё-таки способный взять и определить все имеющиеся белки в протеоме и даже оценить их количество. Речь о шотган-протеоме - где выделенные белки из клетки расщепляются на небольшие фрагменты, разделяются хроматографически и с помощью масс-спектрометрии определяется что это за петид, потому нужно собирать вместе все данные, догадываться какой пептид откуда и пытаться оценить количество. Всё упирается в то, что не все пептиды хорошо летят в приборе и детектируются, и ещё тем, что метод практически не количественный.
Нет нужды напоминать основы биохимии, все помнят что основными компонентами живого являются белки, жиры, нуклеиновые кислоты, вода, неорганические вещества и продукты обмена веществ (метаболизма) как всяческие производные вышеназванного; при этом белки это функциональные исполнители всего-всего, жиры - главный компонент мембран и запас энергии, углеводы используются как носитель/оперативный запас энергии и для конструкционных материалов, нуклеиновые кислоты главным образом являются информационным и регуляторым агентом, вода это основная среда для жизни и неорганические вещества обеспечивают физико-химический "фон" чтобы это всё работало и ещё являются специфическими компонентами для многих ферментов.
Также все помнят основы молекулярной биологии, что ДНК это главное (и единственное) хранилище информации в клетке (хромосомы), изредка используемое как агент переноса генов (плазмиды и прочие векторы); РНК используется как временный носитель информации (мРНК), реже как регулятор чего-нибудь (РНК-интерференция, рибопереключатели) и ещё реже как исполняющий агент (рибозимы) за одним громадным исключением (рибосома); что белки являются главенствующими компонентами живого, которые что-то делают, поскольку делаю они всё - превращение метаболитов, перенос веществ (через мембраны и пр.), детекция веществ, передача сигнала, детекция физико-химических условий, механическую работу, регуляция других компонентов,...
В этом посте мы, после того как вспомнили основной бэкграунд, рассмотрим "центральную догму молекулярной биологии" вообще и что там насчёт контроля, который необходим для управления биологическими процессами и системами.
Это пафосное название, описывающее поток генетической информации, имеющий место (хотя-бы частично) в любом земном живом (или не очень, если речь о вирусах) объекте. Вот картинка:
Закодированная в ДНК информация (геном, вся хромосомная ДНК, геном митохондрий и хлоропластов рассматривают отдельно, плазмиды и торчащие в клетке ДНК-овые вирусы это не_геном) переводится (считывается, транскрибируется) с помощью транскрипции в РНК (транскриптом, всё что транскрибируется с ДНК), если РНК кодирующая (матричная РНК, в ней закодирована последовательность белка, аминокислотная последовательность - КО), то по ней синтезируется (мРНК транслируется) белок (протеом, совокупность всех белков). Схема может быть продолжена, хотя в классическую догму это обычно не включают: совокупность метаболических (упрощая - химических) процессов, производимых в подавляющем большинстве белками, образует метаболом (все метаболические процессы и вещества, в них участвующие), который и реализует фенотип организма (существует редко применямый термин "фенОм", но это затрагивать мы не будем). Вопросы репликации пока поднимать не будем и перейдём к более подробному рассмотрению каждого шага.
Транскрипция
Процесс синтеза РНК по цепи ДНК с помощью РНК-полимеразы (фермента, строящего цепочку из чего-нибудь, здесь из РНК), синтезируются РНК относительно (в смысле, хромосом) небольшой длины на уровне тысяч нт (нуклеотидов), изредка достигая сотен тысяч нт и более. У не_бактерий каждый (нормальный) ген транскрибируется в одной РНК, много генов в одном фрагменте РНК бывает только у бактерий (и вирусов). Модульность "один ген - одна его РНК" (у бактерий часто "одна серия генов - одна их РНК") обеспечивается за счёт наличия более-менее чётких управляющих элементов, техническая аналогия - РНК это ксерокопия чертежа со страницы большой книги. Картина полного цикла транскрипции формального гена:
Транскрипция начинается (инициируется) со специализированных относительно чётких мест в геноме, стоящих недалеко перед геном, они называются "transcription start site" - TSS (да, КО был здесь), транскрипция прекращается (терминируется) на специальных местах остановки, именуемых терминаторами, хотя с ними чёткости меньше. Специальные места в геноме, где располагаются TSS, являются более-менее определёнными последовательностями нуклеотидов - промотерами, точнее промотерными областями генов, с которыми связываются белки, помогающие полимеразам начинать транскрипцию - факторы инициации транскрипции. В процессе исполнения транскрипции (элонгации) нет почти ничего интересного, это опустим. Терминация заканчивается "где-нибудь, главное за геном", потому что этого, чаще всего, достаточно для нормальной работы гена. Важно что с одного куска ДНК (а их всего 1 или 2 на весь геном, исключая повторяющиеся элементы) можно сделать много транскриптов, что обеспечивает "усиление сигнала". Сплайсинг, как фишка только эукариот (об археях не говорим), будет чуть позже.
Трансляция
Процесс синтеза белка по мРНК выполняется с помощью рибосомы (рибозима со вспомогательными белками), использующей тРНК (специальные небольшие РНК с присоединёнными аминокислотами и антикодонами), которая пробегает по мРНК, длины аминокислотных цепей обычно на уровне сотен звеньев. Трансляция выполняется как перевод трёхбуквенных сочетаний нт (кодонов) в аминокислотную последовательность, соответствия тем, что и называется "генетический код". Картинки не будет.
Трансляция инициируется со специального сигнального кодона (стартовго кодона, в общем говоря их несколько, но главный A-U-G), начинать именно с нужного (случайных сочетаний AUG всегда немало) стартового кодона (с которого начинается кодирующая часть РНК) рибосоме помогает контекст (последовательность) и белки (факторы инициации), для разных организмов механизмы отличаются. Цикл элонгации трансляции трогать не будем. Терминация трансляции выполняется за счёт специальных белков на сигнальном кодоне остановки (стоп-кодоне). Опять-же, стоит отметить что по одной мРНК могут пробежать много рибосом, так можно получить много белка с одной молекулы, что обеспечивает "усиление сигнала" и на этапе трансляции.
Последующие процессы уже не имеют обобщённых схем, потому говорить "в общем" уже бессмысленно, перейдём к методам исследований на каждом уровне:
На уровне ДНК - секвенирование (прочтение) геномной ДНК (DNA-seq), уже давно ставшее рутинным (хотя всё ещё дорогостоящим) методом, позволяет получать геном практически полностью для любого объекта живой природы (за исключением некоторых растений), но важно понимать что "прочтение" не значит "понимание" (геномика), одним секвенированием на все вопросы не ответить, потому что нужна информация о том, что именно из себя представляют и как работают все сигнальные и смысловые элементы в последовательности ДНК, а это устанавливается только экспериментами на следующих уровнях. На ранних этапах (первые маленькие геномы в 80-90х годах и большие к 00м) прочтение геномов давало много новых знаний и было "высоконаучно", сейчас (точнее, уже примерно лет 10) это не является передовыми научными исследованиями. Для новых организмов это технический конвейер получения данных (для краткости NGS) для эволюцинистов (и какие-то частности для многих остальных), а для человека это генетические медицинские исследования. В последнем случае это тоже конвейер получения данных для медицинских генетиков, давно являющися рутиной и практически никаких новых открытий не дающий, кроме роста баз сопоставлений генетических вариаций и патологий. Стоит отметить шквалы работ среднего качества по полногеномным исследованиям, в которых оценка влияния обнаруженных геномных вариаций (найденных с помощью методологии "перелопатим всё, найдём что-нибудь") при какой-нибудь патологии не имеет ничего, кроме небольших статистических корреляций, без какой-либо вообще проверки на биологическую адекватность, разбирательства в механизме и проверки на моделях, из-за чего бОльшая часть генетических вариантов в различных базах данных имеет либо никакой эффект (что, обычно, правда), либо слабый эффект, достоверность которого писана вилами на воде. Таким образом, основная сложность "прочитать" вполне преодолена, но она не помогает "понимать". Также это нужно помнить тем, кто говорит "расшифровка генома" (хотя это чисто биоинформатическая работа) заместо "секвенирования генома". Замечание о том, что и сейчас полностью целиком прочитать и собрать геном, например, человека всё ещё не возможно будет описано в следующих постах, а про такое старьё как SNP-чипы я не буду тут рассказывать.
На уровне РНК - измерение количества РНК (для количественной оценки транскрипции как этапа экспресси гена), выполняемое или количественным ПЦР (давно ставшим уже клиническим), если речь по несколько конкретных РНК, либо экспрессионными чипами, если нужно измерять сразу очень много разных известных РНК из известных объектов (ограниченный и устаревающий метод), либо с помощью секвенирования РНК (RNA-seq), которое и прочитает всё, и измерит сколько чего. Собственно в исследованиях на уровне систем клетки хорошо именно последнее, хотя метод дороже чем DNA-seq (который и так дорог), основное его применение - смотреть на то, как меняется транскриптом клетки при каких-либо процессах (воздействия лекарства, вируса, повреждения, дифференциация), т.е. какие гены начали транскрибироваться больше/меньше и ещё что-нибудь, в роде альтернативных сплайс-форм. Позволяет изучать сигнальные каскады, клеточные механизмы и много других важных вещей. Метод тоже хайповый (как и любой ЧегоНибудь-seq), потому есть очень много работ (особенно онкологических, столько, что напрягает читать заголовки журналов в подписках) где любят секвенировать, машинно строить гипотезы о взаимосвязях генов xyz2 и qwe5 при воздействии/патологии H8 на основании статистической корреляции между изменениями экспрессии xyz2 и qwe5 без какого-либо дополнительного разбирательства в механизме. О разных видах RNA-seq и напрягах исследователей будет в специальном после про секвенирование.
Комбинированные методы:
Просто прочтение ДНК и РНК даёт информацию о последовательностях и о их количестве, но это не всё, что имеет место с нуклеиновыми кислотами. С ДНК связываются белки, такие как полимеразы, факторы транскрипции, подавители транскрипции, компоненты систем репарации повреждений (о них будет отдельный пост), белки для репликации, для паковки ДНК (гистоны в случае эукариот), механических манипуляций... и с РНК тоже много чего может связываться (даже больше, РНК гибче), причём связываются эти белки, как правило, с некоторой специфичностью по последовательности (что чаще всего) или структурному мотиву (вспоминаем про комплементарность). Важно знать каков этот мотив последовательности, поскольку это позволяет понимать и предсказывать пути генетической регуляции. Для таких задач применяются методы иммунопреципитации хроматина и РНК (ChIP и RIP), когда ДНК/РНК вместе с белками выделяется, дробится и с помощью антител к интересующему белку вытаскиваются куски ДНК, связавшиеся с нашим белком. Секвенирование этих фрагментов (ChIP-seq и RIP-seq) даёт нам селективное обогащение по кускам ДНК, с которыми связался интересующий белок, так и выясняется где в геноме он садится и каков мотив связывания белка. Для факторов транскрипции, например, это даёт предположения о том, какие гены он может включать.
Вспоминая очень модный нынче тренд "эпигенетика" можно вспомнить не только про ChIP-seq к гистонам (белки, на которые намотана запакованная ДНК в хромосомах) разного вида, но и про химические модификации ДНК (видов которых очень мало, если мы не поливаем клетки ипритом и этиленоксидом), точнее про метилирование. Грубо говоря, метилирование ДНК у эукариот позволяет перманентно запаковать и подавить транскрипцию, перекосы с общим и специфическим метилированим имеют место во многих серьёзных проблемах, таких как старение и рак (потому и хайпа много). С помощью химической модификации (бисульфит обычно, называют BS-seq) можно сделать различимыми при секвенировании метилированные и нет нуклеотиды (технически обработка данных метода значительно труднее и RNA-seq и ChIP-seq, как пришлось как-то убедиться самому). И ещё много инетересных методов, основанных на NGS.
На уровне белка - а вот тут всё плохо, поскольку высокопроизводительного (за счёт массивного параллелизма, как в NGS) метода для секвенирования белков не существует (потому что нет процесса копирования полипептидной цепи, а все работающие секвенаторы сейчас используют именно этот процесс для ДНК). Старые методы разделения белков не справляются с очень многими тысячами белков полной протеомы кого угодно (и в случае высшых эукариот это многообразие осложняется альтернативными формами мРНК), старые методы секвенирования требуют большого количества белка (а неизвестный белок нельзя просто взять и накопировать, в отличии от ДНК и РНК), единственным исключением является метод 2D-GE (за попытку использования этого метода кое-кто может попытаться утопить вас в форезной ванне), которй можно хотя-бы пытаться применять к протеому кого-нибудь, да и то, он сопряжён с масс-спектрометрией... ...на основе которой многие (с некоторым успехом) реализуют метод, всё-таки способный взять и определить все имеющиеся белки в протеоме и даже оценить их количество. Речь о шотган-протеоме - где выделенные белки из клетки расщепляются на небольшие фрагменты, разделяются хроматографически и с помощью масс-спектрометрии определяется что это за петид, потому нужно собирать вместе все данные, догадываться какой пептид откуда и пытаться оценить количество. Всё упирается в то, что не все пептиды хорошо летят в приборе и детектируются, и ещё тем, что метод практически не количественный.