Об обнаружении загрязняющих последовательностей ДНК вектора: повторный анализ необработанных данных
доктора МакКернана.

Статья Др.
Хироси Аракава
В 1991 г. окончил научный факультет Киотского университета. В 1996 г. в Киотском университете получил докторскую степень (молекулярная биология, иммунология).
Работал: Базельский институт иммунологии (Базель)
Институт Генриха Петте (Гамбург)
Институт Гельмгольца (Мюнхен)
Институт Макса Планка (Мюнхен)
Институт молекулярной онкологии (Милан)
Это будет по-прежнему статья о подозрении, что «ДНК подмешана в РНК-коронавирусную вакцину?» Первоначальная цель доктора МакКернана состояла в том, чтобы проверить качество РНК вакцин от Pfizer и Moderna. В процессе он провел комплексный анализ нуклеотидной последовательности РНК вакцины.
Что ж, в этот раз я сам повторно проанализировал необработанные данные доктора и попытался частично воспроизвести процесс.
Глубокое секвенирование также называют NGS (секвенирование следующего поколения). Благодаря распараллеливанию реакций секвенирования эта технология генерирует от нескольких сотен мегабайт до нескольких гигабайт данных базовой последовательности за одну операцию. Технология глубокого секвенирования коренным образом изменила подход к секвенированию генома в науках о жизни.
Давайте посмотрим на фактические данные о базовой последовательности, используя в качестве примера данные о базовой последовательности вакцины Пфайзера. Используемые данные следующие.
Секвенирование двухвалентных мРНК-вакцин Moderna и Pfizer выявило количества от нанограммов до микрограммов двуцепочечной ДНК вектора экспрессии на дозу.
Pfizer Bivalent Vial 1 Прямое считывание
Pfizer Bivalent Vial 1 Обратное считывание
Для получения этих данных д-р МакКернан преобразовал РНК вакцины Pfizer Corona в ДНК и секвенировал ее на секвенаторе Illumina. Ранее я упоминал секвенаторы Illumina в статье об обратной транскрипции . Такие эксперименты называются RNA-seq. Технология глубокого секвенирования включает в себя фрагментацию ДНК, ее амплификацию с помощью ПЦР и параллельное определение последовательности оснований. Технология парного секвенирования определяет последовательность оснований с обоих концов каждого фрагмента ДНК. Даже если последовательность, которую можно прочитать за один раз, короткая, можно анализировать относительно длинные фрагменты ДНК, считывая последовательности нуклеотидов на обоих концах фрагмента ДНК.
С этого момента это будут данные, которые я действительно повторно проанализировал. Для анализа используется приложение CLC Genomics Workbench (версия 23).
Ниже приведен пример последовательности глубокого секвенсора. Последовательность взята из вакцины доктора МакКернана Pfizer. Каждый массив называется чтением. На рисунке ниже показаны пять отведений.

Данные для каждого чтения состоят из имени чтения, последовательности нуклеотидов и показателя качества. Оценка качества означает точность каждой базы. Данные о последовательности такого большого количества операций чтения изначально выводились в виде последовательности букв и цифр в текстовом файле. Приложения для анализа генома визуализируют их.
Глубокое секвенирование включает в себя обработку большого количества прочитанных данных. Данные флакона 1 для вакцины Pfizer составляют 38 561 557 считываний. Количество баз составляет 4 622 741 152 базы. Например, метод фабрикации данных, используемый Министерством здравоохранения, труда и социального обеспечения, представлял собой простую манипуляцию цифрами на графиках. Кроме того, в качестве общей теории иногда говорят о фабрикации данных путем обработки файлов изображений документов, но, напротив, фабриковать данные глубокого секвенирования сложно из-за огромного размера и сложности данных.
Для удобства пояснения в качестве эталонной последовательности используется векторная последовательность (Pbiv1_WM_k141_107), проанализированная доктором МакКернаном . Сравните чтения с векторными последовательностями. Затем, если обнаруживаются почти идентичные последовательности, они накладываются на эту позицию в векторе. Это задача, называемая картированием.
На этот раз мы будем картировать одну последовательность, но также можно картировать весь геном одного вида или транскриптом (сумма всех мРНК).
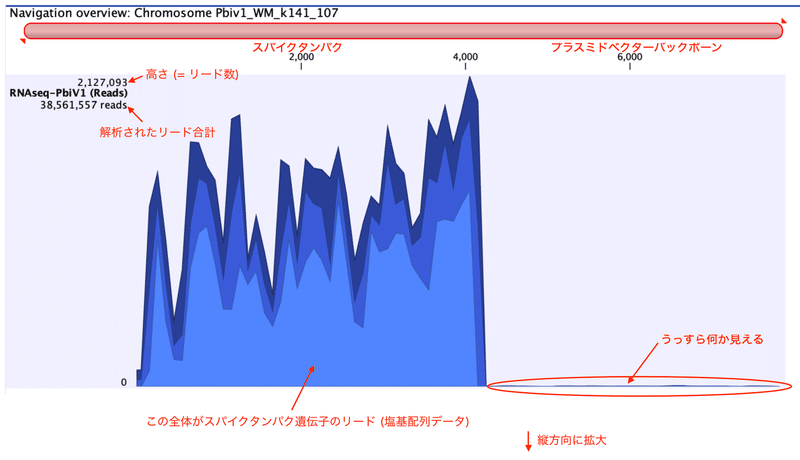
Слева большая гора. Это местонахождение гена спайкового белка. Если присмотреться, в правой части тоже что-то низкое. Высота (количество лидов) на рисунке выше составляет 2 127 093. Давайте отрегулируем высоту и увеличим его. Попробуйте уменьшить высоту до 10000.
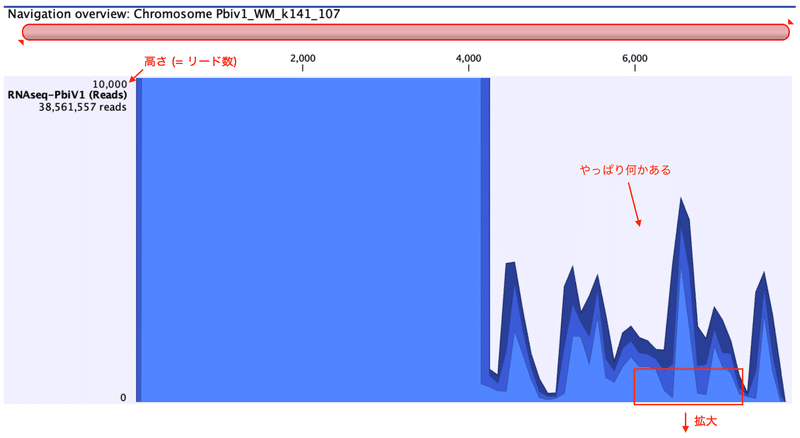
Кажется, что-то есть. Давайте увеличим область, ограниченную прямоугольником выше.
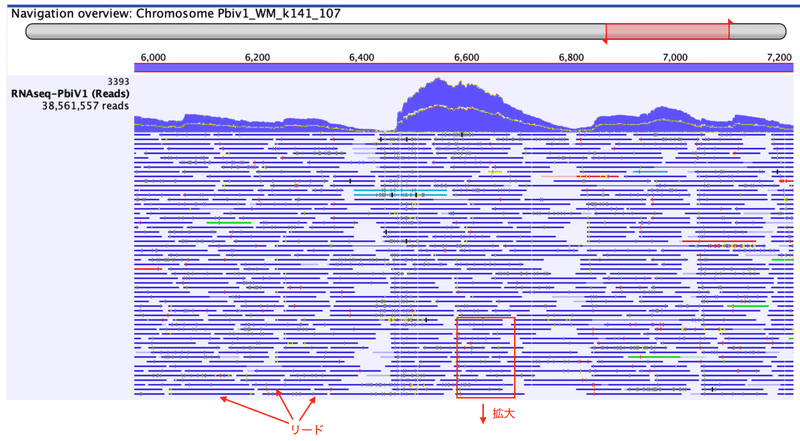
Горизонтальные линии становятся видимыми. Это соответствующие чтения, сопоставленные с соответствующими эталонными последовательностями. Картирование предназначено для сопоставления последовательностей, которые идентичны или очень похожи на эталонную последовательность, с эталонной последовательностью. Давайте увеличим область, ограниченную прямоугольником выше.
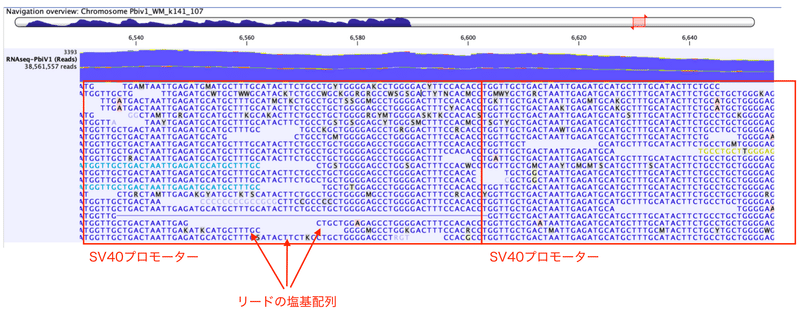
Базовая последовательность каждого чтения теперь видна. Кстати, на этом месте позиция промоутера SV40. Вы можете видеть, что это усиленный тип, который повторяется дважды.
Глубокое секвенирование также представляет собой статистический анализ большого количества нуклеотидных последовательностей. Например, можно получить количественную информацию о том, где в геноме связывается ДНК-связывающий белок. Это техника ChIP-секвенирования. Секвенирование РНК позволяет одновременно сравнивать уровни экспрессии десятков тысяч генов. Даже шаблоны сплайсинга могут быть определены количественно. Одной из сильных сторон глубокого секвенирования является то, что его можно применять к крупномасштабным данным на уровне генома для экспериментов по количественной оценке на уровне одной базы.
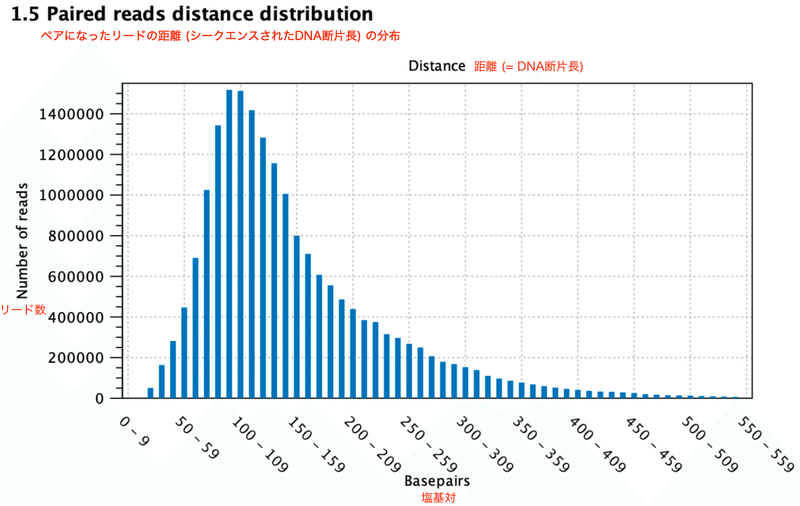
Анализ длины фрагментов ДНК. Распределение длины секвенированных фрагментов ДНК было построено в виде графика. Пиковая длина фрагмента составляет около 100 п.н., а более длинная - около 500 п.н. Если он слишком длинный, ПЦР-амплификация не будет работать эффективно. Если для начала имеется большое количество одноцепочечной ДНК или если ДНК сильно фрагментирована, фрагментация для подготовки образцов для секвенирования еще больше ухудшит ДНК. Такие элементы исключаются из анализа. Здесь мы видим, что фрагменты ДНК, соответствующие остову плазмиды, также были чистыми и имели достаточное качество и длину, чтобы выдержать анализ глубокого секвенирования.
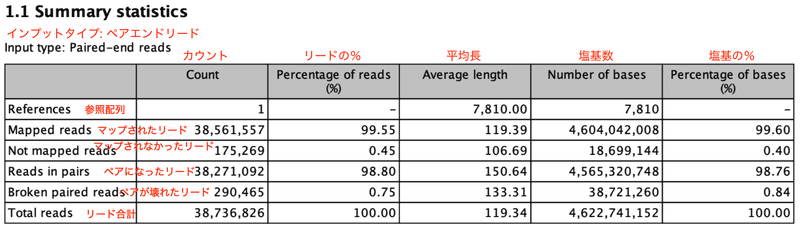
Всего лидов 38 736 826. Они содержат 4 622 741 152 базы данных. Из них 38 561 557 были сопоставлены с эталонными последовательностями. Было нанесено на карту 99,55% от общего количества. Есть также 175 269 не нанесенных на карту отведений. Существуют различные причины, по которым он не был нанесен на карту. Последовательности низкого качества, которые обычно содержат много ошибок секвенирования, не могут быть сопоставлены с эталонными последовательностями и не картированы. Массивы, которые не отображаются, обычно отбрасываются.
Если бы последовательность загрязняющего плазмидного вектора не была известна, как можно было бы вывести последовательность из необработанных данных доктора МакКернана?
Во-первых, считывания последовательностей РНК сравниваются с последовательностями вакцины Pfizer Corona. Другими словами, массив отображается как ссылочный массив. Будет много массивов, которые не отображаются в это время. Так что же это за массив?
Затем извлеките из всего чтения только неотображенные последовательности и сравните их сами с собой. Техника сборки de novo - это методика, которая выравнивает операции чтения без массива ссылок и восстанавливает исходный массив. Затем найдите реконструированную последовательность в банке данных ДНК. Затем появится последовательность остова плазмидного вектора.
Затем снова выровняйте все чтения с помощью сборки de novo без ссылочных массивов. Затем у вас есть последовательность полноразмерной плазмиды с основой плазмидного вектора и геном шиповидного белка.
В общем анализе последовательностей неотображенные последовательности отбрасываются как неинтересные. Если вы посмотрите глубже и не будете игнорировать области, которые обычно упускают из виду, вы можете привести к таким важным открытиям, как открытие доктора МакКернана. Он должен был просто проанализировать обратную транскрипцию ДНК из РНК вакцины против коронавируса, но, возможно, вакцина изначально была заражена плазмидной ДНК? Это он и заметил. На этот раз, благодаря своему собственному анализу, я снова был впечатлен его проницательностью.
Мы планируем дополнительные статьи об этом подозрении на загрязнение ДНК.
https://note.com/hiroshi_arakawa/n/n525d817b16d9
***
Попробую объяснить о чем это и чем опасно.
Несмотря на то, что двухцепочечная ДНК упакована в LNP и содержит последовательность ядерной локализации из промоторов SV40, последовательность не обязательно должна быть локализована в ядре, чтобы возникали проблемы. Цитоплазматическая трансфекция может представлять опасность для делящихся клеток. Во время клеточного деления ядро разбирается и обменивается клеточными компонентами с цитозолем.
Таким образом вам не нужна ядерная локализация для осуществления генетических манипуляций, поскольку ядро будет разбираться в делящихся клетках.
Что еще. Например вопрос о карантине. qPCR C19 поместит вас в карантин для обнаружения sgRNA с CT <40. Это вирусный мусор, обнаруживаемый за пределами слизистого барьера в носу. Загрязнение двухцепочечной ДНК в вакцинах, которые мы наблюдаем, составляет CT 20. Это в 1 миллион раз выше по концентрации, и ее вводят через защиту вашей слизистой оболочки.
Если бы пандемию контролировали с CT <20, пандемии не было бы.
Теперь некоторые будут утверждать, что двухцепочечная ДНК и вирусная РНК являются ложной эквивалентностью, поскольку вирусная РНК способна к репликации. Это неверно. Большинство sgRNA, которые вы обнаруживаете в мазке из носа, НЕ КОМПЕТЕНТНЫ ПО РЕПЛИКАЦИИ, как показано у Jaafar et al. Jaafar et al. Это просто фрагмент РНК, который должен иметь меньшую продолжительность «жизни» в ваших клетках, чем фрагменты, загрязняющие двухцепочечную ДНК. Ну в общем все это ведет к раку как следствие.
Да и еще Pfizer использовал очень нетрадиционный метод секвенирования вакцин. Они использовали метод Oligo Mapping LC/MS, который невозможно распутать и который заваливает рецензента страницами данных и таблиц Mass Spec. Никто не делает этого сегодня для секвенирования ДНК. Данные о секвенировании следующего поколения существуют, но в значительной степени скрыты и представлены только в обобщенном виде. Это сделано намеренно, так как данные следующего поколения выявят загрязнение ДНК, а метод ЖХ/МС - нет. И это сделано намеренно.
МакКернан в частности пишет, что экзонуклеаза Т5 и ДНКаза I (ДНК-нуклеазы) существенно ингибируются присутствием комплементарной мРНК вакцины.
Литература о влиянии m1Ψ на Tm (температура плавления) подразумевает, что m1Ψ является очень «липким» основанием и, вероятно, образует прочно связанные гибриды ДНК-РНК и R-петли, устойчивые к нуклеазам. Это может объяснить, почему плазмидную ДНК было трудно удалить из вакцин. Это также ставит вопрос о том, что делают красители SYBR в присутствии гибридов ДНК/РНК, и находимся ли мы на новой территории, пытаясь количественно определить содержание ДНК в этих вакцинах. Мы упоминали об этом в нашем препринте как о потенциальном ограничении измерений на основе флуорометра.
Это поднимает вопросы относительно того, как клетки млекопитающих воспринимают гибриды ДНК/РНК и являются ли эти гибриды причиной того, что m1Ψ modRNA может быть обнаружена через 28 дней в плазме
Что m1Ψ делает с обработкой R-Loop? Похоже, процессинг R-Loop происходит в пути BRCA1 и является неотъемлемой частью стабильности генома, рака и нейродегенерации.
Статья Др.
Хироси Аракава
В 1991 г. окончил научный факультет Киотского университета. В 1996 г. в Киотском университете получил докторскую степень (молекулярная биология, иммунология).
Работал: Базельский институт иммунологии (Базель)
Институт Генриха Петте (Гамбург)
Институт Гельмгольца (Мюнхен)
Институт Макса Планка (Мюнхен)
Институт молекулярной онкологии (Милан)
Это будет по-прежнему статья о подозрении, что «ДНК подмешана в РНК-коронавирусную вакцину?» Первоначальная цель доктора МакКернана состояла в том, чтобы проверить качество РНК вакцин от Pfizer и Moderna. В процессе он провел комплексный анализ нуклеотидной последовательности РНК вакцины.
Что ж, в этот раз я сам повторно проанализировал необработанные данные доктора и попытался частично воспроизвести процесс.
Глубокое секвенирование также называют NGS (секвенирование следующего поколения). Благодаря распараллеливанию реакций секвенирования эта технология генерирует от нескольких сотен мегабайт до нескольких гигабайт данных базовой последовательности за одну операцию. Технология глубокого секвенирования коренным образом изменила подход к секвенированию генома в науках о жизни.
Давайте посмотрим на фактические данные о базовой последовательности, используя в качестве примера данные о базовой последовательности вакцины Пфайзера. Используемые данные следующие.
Секвенирование двухвалентных мРНК-вакцин Moderna и Pfizer выявило количества от нанограммов до микрограммов двуцепочечной ДНК вектора экспрессии на дозу.
Pfizer Bivalent Vial 1 Прямое считывание
Pfizer Bivalent Vial 1 Обратное считывание
Для получения этих данных д-р МакКернан преобразовал РНК вакцины Pfizer Corona в ДНК и секвенировал ее на секвенаторе Illumina. Ранее я упоминал секвенаторы Illumina в статье об обратной транскрипции . Такие эксперименты называются RNA-seq. Технология глубокого секвенирования включает в себя фрагментацию ДНК, ее амплификацию с помощью ПЦР и параллельное определение последовательности оснований. Технология парного секвенирования определяет последовательность оснований с обоих концов каждого фрагмента ДНК. Даже если последовательность, которую можно прочитать за один раз, короткая, можно анализировать относительно длинные фрагменты ДНК, считывая последовательности нуклеотидов на обоих концах фрагмента ДНК.
С этого момента это будут данные, которые я действительно повторно проанализировал. Для анализа используется приложение CLC Genomics Workbench (версия 23).
Ниже приведен пример последовательности глубокого секвенсора. Последовательность взята из вакцины доктора МакКернана Pfizer. Каждый массив называется чтением. На рисунке ниже показаны пять отведений.
Данные для каждого чтения состоят из имени чтения, последовательности нуклеотидов и показателя качества. Оценка качества означает точность каждой базы. Данные о последовательности такого большого количества операций чтения изначально выводились в виде последовательности букв и цифр в текстовом файле. Приложения для анализа генома визуализируют их.
Глубокое секвенирование включает в себя обработку большого количества прочитанных данных. Данные флакона 1 для вакцины Pfizer составляют 38 561 557 считываний. Количество баз составляет 4 622 741 152 базы. Например, метод фабрикации данных, используемый Министерством здравоохранения, труда и социального обеспечения, представлял собой простую манипуляцию цифрами на графиках. Кроме того, в качестве общей теории иногда говорят о фабрикации данных путем обработки файлов изображений документов, но, напротив, фабриковать данные глубокого секвенирования сложно из-за огромного размера и сложности данных.
Для удобства пояснения в качестве эталонной последовательности используется векторная последовательность (Pbiv1_WM_k141_107), проанализированная доктором МакКернаном . Сравните чтения с векторными последовательностями. Затем, если обнаруживаются почти идентичные последовательности, они накладываются на эту позицию в векторе. Это задача, называемая картированием.
На этот раз мы будем картировать одну последовательность, но также можно картировать весь геном одного вида или транскриптом (сумма всех мРНК).
Слева большая гора. Это местонахождение гена спайкового белка. Если присмотреться, в правой части тоже что-то низкое. Высота (количество лидов) на рисунке выше составляет 2 127 093. Давайте отрегулируем высоту и увеличим его. Попробуйте уменьшить высоту до 10000.
Кажется, что-то есть. Давайте увеличим область, ограниченную прямоугольником выше.
Горизонтальные линии становятся видимыми. Это соответствующие чтения, сопоставленные с соответствующими эталонными последовательностями. Картирование предназначено для сопоставления последовательностей, которые идентичны или очень похожи на эталонную последовательность, с эталонной последовательностью. Давайте увеличим область, ограниченную прямоугольником выше.
Базовая последовательность каждого чтения теперь видна. Кстати, на этом месте позиция промоутера SV40. Вы можете видеть, что это усиленный тип, который повторяется дважды.
Глубокое секвенирование также представляет собой статистический анализ большого количества нуклеотидных последовательностей. Например, можно получить количественную информацию о том, где в геноме связывается ДНК-связывающий белок. Это техника ChIP-секвенирования. Секвенирование РНК позволяет одновременно сравнивать уровни экспрессии десятков тысяч генов. Даже шаблоны сплайсинга могут быть определены количественно. Одной из сильных сторон глубокого секвенирования является то, что его можно применять к крупномасштабным данным на уровне генома для экспериментов по количественной оценке на уровне одной базы.
Анализ длины фрагментов ДНК. Распределение длины секвенированных фрагментов ДНК было построено в виде графика. Пиковая длина фрагмента составляет около 100 п.н., а более длинная - около 500 п.н. Если он слишком длинный, ПЦР-амплификация не будет работать эффективно. Если для начала имеется большое количество одноцепочечной ДНК или если ДНК сильно фрагментирована, фрагментация для подготовки образцов для секвенирования еще больше ухудшит ДНК. Такие элементы исключаются из анализа. Здесь мы видим, что фрагменты ДНК, соответствующие остову плазмиды, также были чистыми и имели достаточное качество и длину, чтобы выдержать анализ глубокого секвенирования.
Всего лидов 38 736 826. Они содержат 4 622 741 152 базы данных. Из них 38 561 557 были сопоставлены с эталонными последовательностями. Было нанесено на карту 99,55% от общего количества. Есть также 175 269 не нанесенных на карту отведений. Существуют различные причины, по которым он не был нанесен на карту. Последовательности низкого качества, которые обычно содержат много ошибок секвенирования, не могут быть сопоставлены с эталонными последовательностями и не картированы. Массивы, которые не отображаются, обычно отбрасываются.
Если бы последовательность загрязняющего плазмидного вектора не была известна, как можно было бы вывести последовательность из необработанных данных доктора МакКернана?
Во-первых, считывания последовательностей РНК сравниваются с последовательностями вакцины Pfizer Corona. Другими словами, массив отображается как ссылочный массив. Будет много массивов, которые не отображаются в это время. Так что же это за массив?
Затем извлеките из всего чтения только неотображенные последовательности и сравните их сами с собой. Техника сборки de novo - это методика, которая выравнивает операции чтения без массива ссылок и восстанавливает исходный массив. Затем найдите реконструированную последовательность в банке данных ДНК. Затем появится последовательность остова плазмидного вектора.
Затем снова выровняйте все чтения с помощью сборки de novo без ссылочных массивов. Затем у вас есть последовательность полноразмерной плазмиды с основой плазмидного вектора и геном шиповидного белка.
В общем анализе последовательностей неотображенные последовательности отбрасываются как неинтересные. Если вы посмотрите глубже и не будете игнорировать области, которые обычно упускают из виду, вы можете привести к таким важным открытиям, как открытие доктора МакКернана. Он должен был просто проанализировать обратную транскрипцию ДНК из РНК вакцины против коронавируса, но, возможно, вакцина изначально была заражена плазмидной ДНК? Это он и заметил. На этот раз, благодаря своему собственному анализу, я снова был впечатлен его проницательностью.
Мы планируем дополнительные статьи об этом подозрении на загрязнение ДНК.
https://note.com/hiroshi_arakawa/n/n525d817b16d9
***
Попробую объяснить о чем это и чем опасно.
Несмотря на то, что двухцепочечная ДНК упакована в LNP и содержит последовательность ядерной локализации из промоторов SV40, последовательность не обязательно должна быть локализована в ядре, чтобы возникали проблемы. Цитоплазматическая трансфекция может представлять опасность для делящихся клеток. Во время клеточного деления ядро разбирается и обменивается клеточными компонентами с цитозолем.
Таким образом вам не нужна ядерная локализация для осуществления генетических манипуляций, поскольку ядро будет разбираться в делящихся клетках.
Что еще. Например вопрос о карантине. qPCR C19 поместит вас в карантин для обнаружения sgRNA с CT <40. Это вирусный мусор, обнаруживаемый за пределами слизистого барьера в носу. Загрязнение двухцепочечной ДНК в вакцинах, которые мы наблюдаем, составляет CT 20. Это в 1 миллион раз выше по концентрации, и ее вводят через защиту вашей слизистой оболочки.
Если бы пандемию контролировали с CT <20, пандемии не было бы.
Теперь некоторые будут утверждать, что двухцепочечная ДНК и вирусная РНК являются ложной эквивалентностью, поскольку вирусная РНК способна к репликации. Это неверно. Большинство sgRNA, которые вы обнаруживаете в мазке из носа, НЕ КОМПЕТЕНТНЫ ПО РЕПЛИКАЦИИ, как показано у Jaafar et al. Jaafar et al. Это просто фрагмент РНК, который должен иметь меньшую продолжительность «жизни» в ваших клетках, чем фрагменты, загрязняющие двухцепочечную ДНК. Ну в общем все это ведет к раку как следствие.
Да и еще Pfizer использовал очень нетрадиционный метод секвенирования вакцин. Они использовали метод Oligo Mapping LC/MS, который невозможно распутать и который заваливает рецензента страницами данных и таблиц Mass Spec. Никто не делает этого сегодня для секвенирования ДНК. Данные о секвенировании следующего поколения существуют, но в значительной степени скрыты и представлены только в обобщенном виде. Это сделано намеренно, так как данные следующего поколения выявят загрязнение ДНК, а метод ЖХ/МС - нет. И это сделано намеренно.
МакКернан в частности пишет, что экзонуклеаза Т5 и ДНКаза I (ДНК-нуклеазы) существенно ингибируются присутствием комплементарной мРНК вакцины.
Литература о влиянии m1Ψ на Tm (температура плавления) подразумевает, что m1Ψ является очень «липким» основанием и, вероятно, образует прочно связанные гибриды ДНК-РНК и R-петли, устойчивые к нуклеазам. Это может объяснить, почему плазмидную ДНК было трудно удалить из вакцин. Это также ставит вопрос о том, что делают красители SYBR в присутствии гибридов ДНК/РНК, и находимся ли мы на новой территории, пытаясь количественно определить содержание ДНК в этих вакцинах. Мы упоминали об этом в нашем препринте как о потенциальном ограничении измерений на основе флуорометра.
Это поднимает вопросы относительно того, как клетки млекопитающих воспринимают гибриды ДНК/РНК и являются ли эти гибриды причиной того, что m1Ψ modRNA может быть обнаружена через 28 дней в плазме
Что m1Ψ делает с обработкой R-Loop? Похоже, процессинг R-Loop происходит в пути BRCA1 и является неотъемлемой частью стабильности генома, рака и нейродегенерации.