Как же далеко до ИИ. Начинаю цикл статей
Предупреждение. Много постов. Они спрятаны. Попасть можно через этот. внизу ссылки
Причина написания постов - на сегодня всё связанное с ИИ -популизмы( МОЙ СЛАБЫЙ ИСКУССТВЕННЫЙ ИНТЕЛЛЕКТ)
, и набор политизированных мемов.
Слишком глубоко вник в тему. Будучи 6 лет вовлечён исключительно в это занятие. Изучил довольно глубоко.
Я специалист по проэктированию приложений Базам Данных. Специализировался на построении онтологий.
Программирую с 1984 года. Интернетом пользуюсь с 1989 года.
Как выглядит пирамида проблематики ИИ.
ИИ.
-----
Экспертные системы.
----
Переводчики языков. Системы распознавания образов. Как сложные -класса гугелмобиля. Так и попроще типа фаинридера.
Распознавание звуков - класса эппла сири. ГЛУБОКОЕ ОБУЧЕНИЕ (DEEP LEARNING) автоматических КОМПОЗИТОРОВ ТЕКСТА Наиболее часто используемые нейронные сети прямого распространения предназначены для обучения с учителем и используются, например, для классификации
Программы игр -шахматы, ГО.
Введение в машинное обучение. Просто о сложном / Хабрахабр
----
Обобщающе -сложное корпоративное ПО. Научные вычисления. Как например хабровские посты по геному. Астрономические программы. Вычисления ядерных и квантовых проблематик.
--------------
Сложные системы БД
Тут и нереляционные БД из мира Интернета, и просто проблематика больших массивов.
-------------------
На сегодня даже основание пирамиды. просто всё связанное с БД - и близко никаких существенных сдвигов. После предложения использовать реляционную алгебру - нет ничего, полный ноль, зеро.
Без онтологичесского описания мира. Невозможно создать компьютерную. Научно целостную модель. Без модели, невозможно создать никаких даже примитивных правил поведения модели. Сответственно исключена возможность создания научно целостного подхода, к наращиванию сложности МетаПравил.
Соответственно и невозможно говорить, ни о каком ИИ.
------------------
Примерчик, для тех кто в теме
5 лет, работал на команды, писавшие для ебея и амазона. занимался исключительно онтологиями. Специализируюсь, с 1999 года, на гетерогенных, распределённых вычислениях. знаю о чём пишу.
простой пример. постройте онтологию геометрических фигур.
1)сначала синтез, потом анализ. от общего к частному.
2)наоборот.
теперь к пункту 1, примените -парадигму наследования.
повторить, с парадигмой реляционной алгебры.
повторить к пункту 2.
--------
1.1
1.2
2.1
2.2
--------
даже на таком примере видно, что конь не валялся.
имеем внешний мир. он описан языком человеческим. оперируется головой.
не имеем для компа, никакого ни внятного описания мира, ни модели. как то сделать.
но уже как мухи об стекло. лет так 45, натягиваем сову на глобус.
ии, эксперт.сис., фортраны изобретаем.
-------------------------
Геномы секвенированных организмов - ошибки в базах / Хабрахабр
30 июня в 03:14Геномы секвенированных организмов - ошибки в базах
Биоинформатика, Алгоритмы*
Наиболее известная база, содержащая геномы секвенированных организмов - NCBI, содержит большое количество систематических ошибок. Из-за этого практически невозможно использование этих данных, и тем более невозможно изучение механизма мутаций (а, следовательно, и эволюции), так как в таком случае исследуются человеческие ошибки при секвенировании, а не природные мутации. Поэтому прежде чем использовать эти данные необходимо уточнение этой базы.
И это трудоемкая задача, её невозможно решить для отдельного нужного организма. Поэтому хотелось бы найти тех, кто хотел бы создать свой русскоязычный источник аналогичный NCBI, но с уточненной информацией.
В статье показывается на сколько массовы ошибки геномов, находящихся в NCBI и рассказывается как самому в этом убедится, и некоторые способы исправления.
Где располагаются геномы
Вот тут на ftp ftp.ncbi.nih.gov/genomes/ расположены все секвенированные геномы. А тут ftp.ncbi.nih.gov/genomes/Bacteria/ геномы бактерий - с них и стоит начать.
Нам нужен файл all.fna.tar.gz - он содержит геномы порядка 2000 бактерий. Что такое геном? Это цепочка ДНК - букв A, T, C, G. Скачиваем, распаковываем - получаем кучу директорий с названиями видов на латинском. Внутри как правило несколько файлов NC_###### - в каждом файле отдельный т.н. локус - нить ДНК (хромосома или плазмида).
Для простоты мы займемся РНК, так как с белками немного посложнее для обработки. Для этого нам нужны еще два файла:
1. all.rnt.tar.gz - содержит список и расположение (начало, конец, направление) всех РНК в определенном локусе
2. all.frn.tar.gz - содержит список собственно вырезанных кусков РНК из ДНК (не удивляйтесь хоть это и РНК - замен T на U здесь не будет - т.к. это именно код ДНК, по которому будет создано РНК)
Как выполнить предобработку
Для обработки данные файлы не очень удобны. Файлы .fna содержат в первой строке комментарий, а далее код ДНК в каждой строке по 70 символов, потом перенос строки. Естественно для поиска это не куда не годится, и нужно это склеить в одну строку без переносов, и исключить комментарий. Обработанному таким образом файлу дадим расширение .fna.txt
Кроме того, есть еще один нюанс: с ДНК может осуществляться транскрипция РНК как справа налево, так и наоборот, а так как ДНК - это у нас двойная спираль, то справа налево означает транскрипцию с одной нити ДНК, а слева направо с другой комплементарной ей.
Это означает, что чтобы найти к примеру РНК для которой указано отрицательное направление в файле all.rnt.tar.gz - нам нужно искать не в полученном файле .fna.txt - так мы ничего не найдем. Нужно создать обратный файл (дадим ему расширение .fna_.txt). Суть там такая берем файл .fna.txt - читаем побуквенно с конца и делаем комплементарные замены:
T=A; G=C; A=T; C=G; M=K; R=Y; W=W; S=S; Y=R; K=M; V=B; H=D; D=H; B=V
Первых 4 понятны и известны. Остальные достаточно неожиданны :), правда они встречаются редко, но встречаются. Суть в них такая если при секвенировании невозможно отличить G от A ставится R и т.п.
Находим ошибки или кросс-анализ
Этому процессу я дал название кросс-анализ. Суть такая из интересующего файла .frn берем код одной РНК. И ищем совпадение по всему множеству файлов .fna.txt и .fna_.txt.
Как вы думаете сколько вы получите совпадений? Достаточно. Причем может оказаться, что в файле .rnt может не оказаться соответствующей записи. А наиболее часто окажется, что начало и конец в файле будет не таким, а сдвинутым на 1 или 3 позиции. Может даже быть другое направление. Встречался я и с ошибками более существенными когда указано, что это Ile РНК, а на самом деле это Met РНК.
По числу этих ошибок практически порядка 50%. Как можно работать с такими ошибочными данными я не знаю. Почему люди из NCBI не проводят такой простой кросс-анализ - тоже не знаю.
Но прикиньте сколько ошибочных выводов потом делают биологи доверившись этим данным?
В то же время, когда ошибки исправлены - этот же метод позволяет сделать эксперимент такого типа, как описан тут Интересные результаты о эволюционной систематике прокариот или «многовидовое происхождение», отдельно отметим на сколько это элементарный метод, но дающий точные факты, которые могут говорить о многом.
Один самый простой пример
Открываем секвенированный организм Chlamydophila pneumoniae TW-183. Поиском ищем тег «CpBt08», там указано complement(266485..266557) - это начало и конец соответственно. Там же есть ссылка наGeneID:3284349. Далее есть ссылка FASTA - там указана последовательность
CGGGGACTTAGCTTAGTTGGTAGAGCGTCTGATTTGCATTCAGAAGGTCAGGAGTTCGAATCTCCTAGTCTCC
она не верная - на самом деле должна быть
GGGGACTTAGCTTAGTTGGTAGAGCGTCTGATTTGCATTCAGAAGGTCAGGAGTTCGAATCTCCTAGTCTCCA
(и она действительно такая есть в полном секвенсе ДНК, она лишь неправильно идентифицирована)
поставим их рядом
CGGGGACTTAGCTTAGTTGGTAGAGCGTCTGATTTGCATTCAGAAGGTCAGGAGTTCGAATCTCCTAGTCTCC GGGGACTTAGCTTAGTTGGTAGAGCGTCTGATTTGCATTCAGAAGGTCAGGAGTTCGAATCTCCTAGTCTCCA
видим что отличие в сдвиге.
Почему?
Теперь идем в другой организм Chlamydophila pneumoniae CWL029, ищем тег: CPnt08. И аналогично находим ген
GGGGACTTAGCTTAGTTGGTAGAGCGTCTGATTTGCATTCAGAAGGTCAGGAGTTCGAATCTCCTAGTCTCCA
вы думаете это просто другая последовательность? Ан, нет - это та же сама но сдвинутая. Конечно есть, вопрос какая из них правильная. И тут самое плохое - автоматически это решить затруднительно. Приходится решать основываясь на частоты ошибок и некоторого знания о том какими могут быть последовательности РНК. Впрочем, для тРНК есть более конкретный критерий проверить соответствие антикодона в позициях 34-36 и наличии конца ССА (который должен быть у всех тРНК).
P.S. Кого это заинтересует настолько, чтобы попробовать поискать ошибки, исправить или даже сделать эксперимент аналогичный моему, но на других данных - обращайтесь личным сообщением.--------------------------------------
Интересные результаты о эволюционной систематике прокариот или «многовидовое происхождение» / Хабрахабр
28 июня в 16:43Интересные результаты о эволюционной систематике прокариот или «многовидовое происхождение»
Биоинформатика, Алгоритмы*
Филогенетическая систематика пытается определить родство различных организмов и их эволюционную близость. Если не так давно об этом судили по внешним признакам организмов (морфологии если точнее), то теперь однозначно перешли к суждению путем сравнения геномов этих организмов.
Но ДНК организма состоит из множества нуклеотидов и учитывать их все для определения схожести организмов сильно затруднительно. Кроме того ДНК постоянно эволюционирует. Поэтому биологи начали основываться на рибосомной рибонуклеиновой кислоте (рРНК), т.к. эти молекулы обнаружены у всех клеточных форм жизни, их функции связаны с важнейшим для организма процессом трансляции, первичная структура в целом характеризуется высокой консервативностью.
Считается, что особенностью рРНК является нахождение вне сферы действия отбора, поэтому данные молекулы эволюционируют в результате спонтанных мутаций, происходящих с постоянной скоростью, и накопление таких мутаций зависит только от времени. Таким образом, мерой эволюционного расстояния между организмами служит количество нуклеотидных замен в молекулах сравниваемых рРНК.
Известно, что в рибосомах прокариот и эукариот присутствуют 3 типа рРНК. Информационная емкость крупных молекул больше, но их труднее анализировать. Поэтому наиболее удобным оказался анализ молекул рРНК средней величины: 16S (~1600 нуклеотидов). Систематика основывается на расчете коэффициентов сходства сравниваемых организмов. Именно на основании анализа рРНК современная систематика выделяет три домена бактерии, археи и эукариоты, а так же на этом основывается систематика, бактерий и архей X издания Берги.
Вот такое положение дел в этой сфере на данный момент. Мной же была сделана попытка создать основы для несколько другой, если хотите альтернативной, систематики. Почему? Консервативность рРНК тем не менее не достаточно велика, консервативны лишь некоторые её части. А так как есть достаточно вариабельные части у рРНК, то приходится делать допущения и предполагать, где были разрывы и вставки отдельных фрагментов при мутации. А т.н. выравнивание сейчас делается с очень большой погрешностью.
В итоге, я пришел к выводу, что необходимо при сравнении геномных последовательностей сравнивать такие участки, которые вообще не подвергались мутациям, и которые абсолютно идентичны в разных организмах.
Смотрим, что из этого получилось.
Существуют ли участки ДНК, которые вообще не подвергаются мутациям на протяжении длительного времени?
Но есть ли такие участки, которые вообще не подвергались мутациям, и абсолютно идентичны в разных организмах? Оказывается есть. Целый ряд белков (их ДНК код) абсолютно одинаковы для многих видов, отнесенных к одному роду, или даже семейству. Но еще большей консервативностью обладают транспортные РНК (тРНК). В хромосоме бактерии, как правило, существуют все 20 видов тРНК, каждая из которых ответственна за транспортировку определенной аминокислоты к месту синтеза белка. И вот на основании их можно проследить эволюционную связь не только отдельных семейств, но и целых классов и даже типов.
В данном исследовании я основывался только на одной тРНК, которая транспортирует аланин (Ala tRNA). Поэтому выводы о эволюционной связи не претендуют на окончательный результат. Для этого нужно сопоставить результаты в отношении других тРНК. Но тем не менее, проделанное исследование позволяет наметить ряд положений о близости некоторых родов бактерий. Затем будет показано, что о близких эволюционных связях нельзя говорить в терминах филогенетического дерева. Это вызвано тем фактом, что существует горизонтальный перенос и конъюгация у бактерий, и тут требуются другие подходы.
Анализ геномов
В центре нашего внимания будет род Yersinia (Чумная палочка), но в результате исследования были рассмотрены и другие такие как Shewanella, Pseudomonas, Vibrio, Erythrobacter, Pseudoalteromonas, Photobacterium и ряд других (всего 109 локусов)
тРНК у бактерий, как правило, имеет константную длину 76 нуклеотидов, при этом антикодон располагается в позициях 34,35,36. Аланин в ДНК кодируется четырьмя кодонами: GCT, GCC, GCA, GCG. Поэтому потенциально возможны 4 вида Ala tRNA, с антикодонами AGC, GGC, TGC, CGC.
Но у подавляющего большинства рассматриваемых бактерий в геноме есть только по 2 вида Ala tRNA_GCA и Ala tRNA_GCС. Есть, конечно, и исключения.
Для анализа были использованы секвенированные геномы ДНК, которые имеются в базе NCBI. Все идентичные немутировавшие тРНК были помечены уникальным идентификатором (Id). С помощь написанной для анализа компьютерной программы, в полуавтоматическом режиме с ручной верификацией, был составленсписок разнообразных видов Ala tRNA, и их расположение в том или ином секвенированном локусе.
Результаты
У всех рассматриваемых штаммов рода Yersinia (9 шт.), имеется в геноме одинаковые Ala tRNA_GCA с Id=00046 и Ala tRNA_GCС с Id=00043. На основании этого факта, можно сделать вывод, что действительно эти штаммы имеют сильную эволюционную связь, и поэтому их всех относят к роду Yersinia.
Сейчас род Yersinia относят к семейству Enterobacteriaceae. Но на основании проделанного анализа, в рамках сходства по Ala tRNA, эта связь малообоснованна.
Если посмотреть на классических представителей семейства Enterobacteriaceae, таких как Escherichia, Salmonella, Shigella, Citrobacter, Cronobacter, Klebsiella, Pectobacterium, то у всех них совершенно другие Ala tRNA. А именно у них Ala tRNA_GCA с Id=00011 и Ala tRNA_GCС с Id=00012. На этом основании мы и можем считать перечисленные роды семейства Enterobacteriaceae классическими представителями.
И только с родом Photorhabdus, по одной линии Ala tRNA_GCA с Id=00046, Yersinia имеет связь. Поэтому род Yersinia имеет черты от разных семейств. Такой род мы назовем переходным родом между различными семействами.
Итак, если с семейством Enterobacteriaceae род Yersinia связывает только одна конфигурация не мутировавшей Ala tRNA_GCA с Id=00046 (и то частично), то встает вопрос с каким семейством род Yersinia связан по другой конфигурации не мутировавшей Ala tRNA_GCС с Id=00043?
Оказывается наиболее прямой связью по этой линии является род Shewanella (семейство: Shewanellaceae, порядок: Alteromonadales, класс: гамма-протеобактерии). При этом объединяющая их линия Ala tRNA_GCС с Id=00043, является ключевой в эволюционном плане, так как присутствует так же еще у родов Pseudomonas и Vibrio. Все эти связи более близкие, чем следует из современной классификации, где эти роды объединены лишь на уровне класса.
В свою очередь некоторые представители рода Shewanella имеют одинаковые Ala tRNA_GCA с Id=00047 и Ala tRNA_GCС с Id=00043. Уже выяснив, что Yersinia и Shewanella имеют связь по одной линии (Id=00043), интересно с кем связан род Shewanella по другой линии (Id=00047). Оказывается, что эта линия также является достаточно ключевой в эволюционном плане. Она разветвляет происхождение далее и Ala tRNA_GCA с Id=00047 обладают так же представители родов Vibrio, Thiomicrospira, Saccharophagus.
Мы можем также проследить эти связи и далее (см. рисунок). Но уже сейчас понятно, чтобы систематизировать эти эволюционные связи, нужно несколько изменить подход к их описанию.
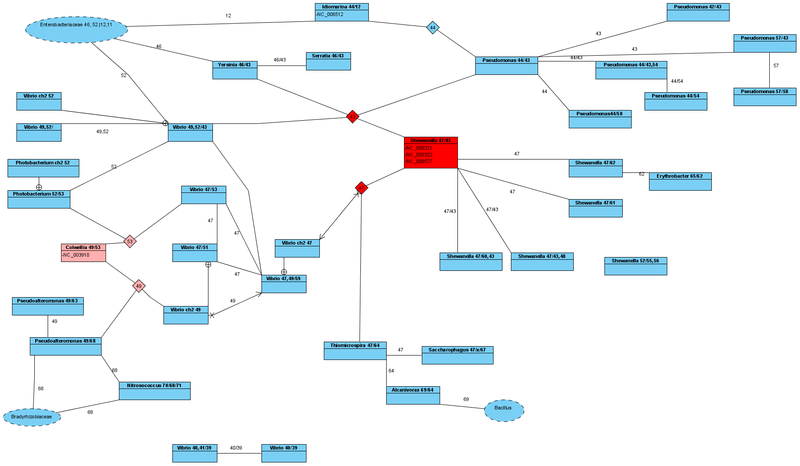
в более высоком разрешении
Выводы
Следует еще раз подчеркнуть, что все выводы сделанные в работе, базируются на анализе только tRNA Ala, и конечно для признания результатов нужна проверка по другим видам тРНК. Но тем ни менее уже сейчас мы можем сделать некоторые выводы и описать каким образом нужно изменить подходы к систематике таксонов.
Достаточную сложность вызывает определение какой из видов более эволюционно древний, а какой более молодой. Но если исходить из гипотезы, что биологический мир развивался от простого к сложному, то у нас есть по крайней мере один неоспоримый факт. Если бактерия имеет две хромосомы, то кажется очевидным, что она эволюционно более молода, чем имеющая одну.
Поэтому в рамках нашего исследования можно с уверенностью сказать, что род Vibrio более молодой, чем Shewanella или Yersinia. И тогда, раз Vibrio и Shewanella объединены идентичными Ala tRNAGCA с Id=00047, то с большой вероятностью, род Vibrio произошел от Shewanella и так произошла одна хромосома. После чего другие представители рода Vibrio произошли от рода Colwellia и так произошла вторая хромосома. В результате объединения этих хромосом в одном организме мы и можем говорить о роде Vibrio, который по одной линии произошел от Shewanella, а по другой от Colwellia.
Таким образом, мы должны говорить не о происхождении от одного предка, а о как минимум двух, а то и больше.
С однохромосомными бактериями сложнее определить направление эволюции (кто более молодой, а кто более древний). Но основываясь на двуххромосомные виды, можно сказать, так как существует вид Vibrio с Ala tRNA_GCA с Id=00049 и Ala tRNA_GCС с Id=00043; а также существуют виды Vibrio с Ala tRNA_GCA с Id=00047, то первоночально существовали именно Ala tRNA_GCA с Id=00047 и Ala tRNA_GCС с Id=00043. А они содержались в Shewanella, и поэтому его нужно признать наиболее древним организмом, и положить в основу систематики рассмотренных здесь организмов.
Тогда можно сделать вывод, что от Shewanella по одной линии произошли Yersinia. От Yersinia в свою очередь Photorhabdus, от которого и произошло все семейство Enterobacteriaceae. Но это только по одной линии. По другой же мы уже упоминали какие роды произошли от Shewanella.
Многовидовое происхождение сильно запутывает эволюционную картину, но с этим ничего не поделаешь - такова сложность видообразования, и нам нужно лишь их наиболее точно отразить в условиях, когда не все виды известны.
upd. Ах, да. Совсем забыл, тут начали говорить - ну зачем программистам дурить голову всякой биологией. Так я собственно, и хотел заинтересовать именно программистов этой темой, так как именно они способны написать алгоритмы для биовычислений. У меня просто у самого не хватает сил провести более полный анализ. Вдруг кого-то заинтересует милости прошу пишите в личку.----------------
КАК ЖЕ ДАЛЕКО ДО ИИ. КАМЕНТЫ
КАК ЖЕ ДАЛЕКО ДО ИИ. КАМЕНТЫ2
КАК ЖЕ ДАЛЕКО ДО ИИ. КАМЕНТЫ3
--------------
отрывок
http://evolbiol.ru/latgentrans.htm
Горизонтальный перенос генов и его роль в эволюции
Долгое время эволюционная теория базировалась на представлении о том, что виды не могут обмениваться друг с другом наследственной информацией. Как только вид делится на два, и между ними возникает репродуктивная изоляция, они эволюционируют дальше сами по себе по схеме "случайные мутации + естественный отбор". На этом основывалась и Дарвиновская схема дивергенции, и столь модный на Западе кладизм - "филогенетическая систематика", и все "эволюционные деревья" и иные реконструкции исторического развития биоты. Однако расчеты показывали, что при такой изолированной эволюции отдельных видов на основе случайных мутаций и отбора жизнь просто не успела бы за сравнительно недолгий срок своего существования (4 млрд. лет) развиться от простейших форм до таких высокоорганизованных, как млекопитающие и человек.
-----------------------------
КАК ЖЕ ДАЛЕКО ДО ИИ. НЦИКЛ СТАТЕЙ 2
Причина написания постов - на сегодня всё связанное с ИИ -популизмы( МОЙ СЛАБЫЙ ИСКУССТВЕННЫЙ ИНТЕЛЛЕКТ)
, и набор политизированных мемов.
Слишком глубоко вник в тему. Будучи 6 лет вовлечён исключительно в это занятие. Изучил довольно глубоко.
Я специалист по проэктированию приложений Базам Данных. Специализировался на построении онтологий.
Программирую с 1984 года. Интернетом пользуюсь с 1989 года.
Как выглядит пирамида проблематики ИИ.
ИИ.
-----
Экспертные системы.
----
Переводчики языков. Системы распознавания образов. Как сложные -класса гугелмобиля. Так и попроще типа фаинридера.
Распознавание звуков - класса эппла сири. ГЛУБОКОЕ ОБУЧЕНИЕ (DEEP LEARNING) автоматических КОМПОЗИТОРОВ ТЕКСТА Наиболее часто используемые нейронные сети прямого распространения предназначены для обучения с учителем и используются, например, для классификации
Программы игр -шахматы, ГО.
Введение в машинное обучение. Просто о сложном / Хабрахабр
----
Обобщающе -сложное корпоративное ПО. Научные вычисления. Как например хабровские посты по геному. Астрономические программы. Вычисления ядерных и квантовых проблематик.
--------------
Сложные системы БД
Тут и нереляционные БД из мира Интернета, и просто проблематика больших массивов.
-------------------
На сегодня даже основание пирамиды. просто всё связанное с БД - и близко никаких существенных сдвигов. После предложения использовать реляционную алгебру - нет ничего, полный ноль, зеро.
Без онтологичесского описания мира. Невозможно создать компьютерную. Научно целостную модель. Без модели, невозможно создать никаких даже примитивных правил поведения модели. Сответственно исключена возможность создания научно целостного подхода, к наращиванию сложности МетаПравил.
Соответственно и невозможно говорить, ни о каком ИИ.
------------------
Примерчик, для тех кто в теме
5 лет, работал на команды, писавшие для ебея и амазона. занимался исключительно онтологиями. Специализируюсь, с 1999 года, на гетерогенных, распределённых вычислениях. знаю о чём пишу.
простой пример. постройте онтологию геометрических фигур.
1)сначала синтез, потом анализ. от общего к частному.
2)наоборот.
теперь к пункту 1, примените -парадигму наследования.
повторить, с парадигмой реляционной алгебры.
повторить к пункту 2.
--------
1.1
1.2
2.1
2.2
--------
даже на таком примере видно, что конь не валялся.
имеем внешний мир. он описан языком человеческим. оперируется головой.
не имеем для компа, никакого ни внятного описания мира, ни модели. как то сделать.
но уже как мухи об стекло. лет так 45, натягиваем сову на глобус.
ии, эксперт.сис., фортраны изобретаем.
-------------------------
Геномы секвенированных организмов - ошибки в базах / Хабрахабр
30 июня в 03:14Геномы секвенированных организмов - ошибки в базах
Биоинформатика, Алгоритмы*
Наиболее известная база, содержащая геномы секвенированных организмов - NCBI, содержит большое количество систематических ошибок. Из-за этого практически невозможно использование этих данных, и тем более невозможно изучение механизма мутаций (а, следовательно, и эволюции), так как в таком случае исследуются человеческие ошибки при секвенировании, а не природные мутации. Поэтому прежде чем использовать эти данные необходимо уточнение этой базы.
И это трудоемкая задача, её невозможно решить для отдельного нужного организма. Поэтому хотелось бы найти тех, кто хотел бы создать свой русскоязычный источник аналогичный NCBI, но с уточненной информацией.
В статье показывается на сколько массовы ошибки геномов, находящихся в NCBI и рассказывается как самому в этом убедится, и некоторые способы исправления.
Где располагаются геномы
Вот тут на ftp ftp.ncbi.nih.gov/genomes/ расположены все секвенированные геномы. А тут ftp.ncbi.nih.gov/genomes/Bacteria/ геномы бактерий - с них и стоит начать.
Нам нужен файл all.fna.tar.gz - он содержит геномы порядка 2000 бактерий. Что такое геном? Это цепочка ДНК - букв A, T, C, G. Скачиваем, распаковываем - получаем кучу директорий с названиями видов на латинском. Внутри как правило несколько файлов NC_###### - в каждом файле отдельный т.н. локус - нить ДНК (хромосома или плазмида).
Для простоты мы займемся РНК, так как с белками немного посложнее для обработки. Для этого нам нужны еще два файла:
1. all.rnt.tar.gz - содержит список и расположение (начало, конец, направление) всех РНК в определенном локусе
2. all.frn.tar.gz - содержит список собственно вырезанных кусков РНК из ДНК (не удивляйтесь хоть это и РНК - замен T на U здесь не будет - т.к. это именно код ДНК, по которому будет создано РНК)
Как выполнить предобработку
Для обработки данные файлы не очень удобны. Файлы .fna содержат в первой строке комментарий, а далее код ДНК в каждой строке по 70 символов, потом перенос строки. Естественно для поиска это не куда не годится, и нужно это склеить в одну строку без переносов, и исключить комментарий. Обработанному таким образом файлу дадим расширение .fna.txt
Кроме того, есть еще один нюанс: с ДНК может осуществляться транскрипция РНК как справа налево, так и наоборот, а так как ДНК - это у нас двойная спираль, то справа налево означает транскрипцию с одной нити ДНК, а слева направо с другой комплементарной ей.
Это означает, что чтобы найти к примеру РНК для которой указано отрицательное направление в файле all.rnt.tar.gz - нам нужно искать не в полученном файле .fna.txt - так мы ничего не найдем. Нужно создать обратный файл (дадим ему расширение .fna_.txt). Суть там такая берем файл .fna.txt - читаем побуквенно с конца и делаем комплементарные замены:
T=A; G=C; A=T; C=G; M=K; R=Y; W=W; S=S; Y=R; K=M; V=B; H=D; D=H; B=V
Первых 4 понятны и известны. Остальные достаточно неожиданны :), правда они встречаются редко, но встречаются. Суть в них такая если при секвенировании невозможно отличить G от A ставится R и т.п.
Находим ошибки или кросс-анализ
Этому процессу я дал название кросс-анализ. Суть такая из интересующего файла .frn берем код одной РНК. И ищем совпадение по всему множеству файлов .fna.txt и .fna_.txt.
Как вы думаете сколько вы получите совпадений? Достаточно. Причем может оказаться, что в файле .rnt может не оказаться соответствующей записи. А наиболее часто окажется, что начало и конец в файле будет не таким, а сдвинутым на 1 или 3 позиции. Может даже быть другое направление. Встречался я и с ошибками более существенными когда указано, что это Ile РНК, а на самом деле это Met РНК.
По числу этих ошибок практически порядка 50%. Как можно работать с такими ошибочными данными я не знаю. Почему люди из NCBI не проводят такой простой кросс-анализ - тоже не знаю.
Но прикиньте сколько ошибочных выводов потом делают биологи доверившись этим данным?
В то же время, когда ошибки исправлены - этот же метод позволяет сделать эксперимент такого типа, как описан тут Интересные результаты о эволюционной систематике прокариот или «многовидовое происхождение», отдельно отметим на сколько это элементарный метод, но дающий точные факты, которые могут говорить о многом.
Один самый простой пример
Открываем секвенированный организм Chlamydophila pneumoniae TW-183. Поиском ищем тег «CpBt08», там указано complement(266485..266557) - это начало и конец соответственно. Там же есть ссылка наGeneID:3284349. Далее есть ссылка FASTA - там указана последовательность
CGGGGACTTAGCTTAGTTGGTAGAGCGTCTGATTTGCATTCAGAAGGTCAGGAGTTCGAATCTCCTAGTCTCC
она не верная - на самом деле должна быть
GGGGACTTAGCTTAGTTGGTAGAGCGTCTGATTTGCATTCAGAAGGTCAGGAGTTCGAATCTCCTAGTCTCCA
(и она действительно такая есть в полном секвенсе ДНК, она лишь неправильно идентифицирована)
поставим их рядом
CGGGGACTTAGCTTAGTTGGTAGAGCGTCTGATTTGCATTCAGAAGGTCAGGAGTTCGAATCTCCTAGTCTCC GGGGACTTAGCTTAGTTGGTAGAGCGTCTGATTTGCATTCAGAAGGTCAGGAGTTCGAATCTCCTAGTCTCCA
видим что отличие в сдвиге.
Почему?
Теперь идем в другой организм Chlamydophila pneumoniae CWL029, ищем тег: CPnt08. И аналогично находим ген
GGGGACTTAGCTTAGTTGGTAGAGCGTCTGATTTGCATTCAGAAGGTCAGGAGTTCGAATCTCCTAGTCTCCA
вы думаете это просто другая последовательность? Ан, нет - это та же сама но сдвинутая. Конечно есть, вопрос какая из них правильная. И тут самое плохое - автоматически это решить затруднительно. Приходится решать основываясь на частоты ошибок и некоторого знания о том какими могут быть последовательности РНК. Впрочем, для тРНК есть более конкретный критерий проверить соответствие антикодона в позициях 34-36 и наличии конца ССА (который должен быть у всех тРНК).
P.S. Кого это заинтересует настолько, чтобы попробовать поискать ошибки, исправить или даже сделать эксперимент аналогичный моему, но на других данных - обращайтесь личным сообщением.--------------------------------------
Интересные результаты о эволюционной систематике прокариот или «многовидовое происхождение» / Хабрахабр
28 июня в 16:43Интересные результаты о эволюционной систематике прокариот или «многовидовое происхождение»
Биоинформатика, Алгоритмы*
Филогенетическая систематика пытается определить родство различных организмов и их эволюционную близость. Если не так давно об этом судили по внешним признакам организмов (морфологии если точнее), то теперь однозначно перешли к суждению путем сравнения геномов этих организмов.
Но ДНК организма состоит из множества нуклеотидов и учитывать их все для определения схожести организмов сильно затруднительно. Кроме того ДНК постоянно эволюционирует. Поэтому биологи начали основываться на рибосомной рибонуклеиновой кислоте (рРНК), т.к. эти молекулы обнаружены у всех клеточных форм жизни, их функции связаны с важнейшим для организма процессом трансляции, первичная структура в целом характеризуется высокой консервативностью.
Считается, что особенностью рРНК является нахождение вне сферы действия отбора, поэтому данные молекулы эволюционируют в результате спонтанных мутаций, происходящих с постоянной скоростью, и накопление таких мутаций зависит только от времени. Таким образом, мерой эволюционного расстояния между организмами служит количество нуклеотидных замен в молекулах сравниваемых рРНК.
Известно, что в рибосомах прокариот и эукариот присутствуют 3 типа рРНК. Информационная емкость крупных молекул больше, но их труднее анализировать. Поэтому наиболее удобным оказался анализ молекул рРНК средней величины: 16S (~1600 нуклеотидов). Систематика основывается на расчете коэффициентов сходства сравниваемых организмов. Именно на основании анализа рРНК современная систематика выделяет три домена бактерии, археи и эукариоты, а так же на этом основывается систематика, бактерий и архей X издания Берги.
Вот такое положение дел в этой сфере на данный момент. Мной же была сделана попытка создать основы для несколько другой, если хотите альтернативной, систематики. Почему? Консервативность рРНК тем не менее не достаточно велика, консервативны лишь некоторые её части. А так как есть достаточно вариабельные части у рРНК, то приходится делать допущения и предполагать, где были разрывы и вставки отдельных фрагментов при мутации. А т.н. выравнивание сейчас делается с очень большой погрешностью.
В итоге, я пришел к выводу, что необходимо при сравнении геномных последовательностей сравнивать такие участки, которые вообще не подвергались мутациям, и которые абсолютно идентичны в разных организмах.
Смотрим, что из этого получилось.
Существуют ли участки ДНК, которые вообще не подвергаются мутациям на протяжении длительного времени?
Но есть ли такие участки, которые вообще не подвергались мутациям, и абсолютно идентичны в разных организмах? Оказывается есть. Целый ряд белков (их ДНК код) абсолютно одинаковы для многих видов, отнесенных к одному роду, или даже семейству. Но еще большей консервативностью обладают транспортные РНК (тРНК). В хромосоме бактерии, как правило, существуют все 20 видов тРНК, каждая из которых ответственна за транспортировку определенной аминокислоты к месту синтеза белка. И вот на основании их можно проследить эволюционную связь не только отдельных семейств, но и целых классов и даже типов.
В данном исследовании я основывался только на одной тРНК, которая транспортирует аланин (Ala tRNA). Поэтому выводы о эволюционной связи не претендуют на окончательный результат. Для этого нужно сопоставить результаты в отношении других тРНК. Но тем не менее, проделанное исследование позволяет наметить ряд положений о близости некоторых родов бактерий. Затем будет показано, что о близких эволюционных связях нельзя говорить в терминах филогенетического дерева. Это вызвано тем фактом, что существует горизонтальный перенос и конъюгация у бактерий, и тут требуются другие подходы.
Анализ геномов
В центре нашего внимания будет род Yersinia (Чумная палочка), но в результате исследования были рассмотрены и другие такие как Shewanella, Pseudomonas, Vibrio, Erythrobacter, Pseudoalteromonas, Photobacterium и ряд других (всего 109 локусов)
тРНК у бактерий, как правило, имеет константную длину 76 нуклеотидов, при этом антикодон располагается в позициях 34,35,36. Аланин в ДНК кодируется четырьмя кодонами: GCT, GCC, GCA, GCG. Поэтому потенциально возможны 4 вида Ala tRNA, с антикодонами AGC, GGC, TGC, CGC.
Но у подавляющего большинства рассматриваемых бактерий в геноме есть только по 2 вида Ala tRNA_GCA и Ala tRNA_GCС. Есть, конечно, и исключения.
Для анализа были использованы секвенированные геномы ДНК, которые имеются в базе NCBI. Все идентичные немутировавшие тРНК были помечены уникальным идентификатором (Id). С помощь написанной для анализа компьютерной программы, в полуавтоматическом режиме с ручной верификацией, был составленсписок разнообразных видов Ala tRNA, и их расположение в том или ином секвенированном локусе.
Результаты
У всех рассматриваемых штаммов рода Yersinia (9 шт.), имеется в геноме одинаковые Ala tRNA_GCA с Id=00046 и Ala tRNA_GCС с Id=00043. На основании этого факта, можно сделать вывод, что действительно эти штаммы имеют сильную эволюционную связь, и поэтому их всех относят к роду Yersinia.
Сейчас род Yersinia относят к семейству Enterobacteriaceae. Но на основании проделанного анализа, в рамках сходства по Ala tRNA, эта связь малообоснованна.
Если посмотреть на классических представителей семейства Enterobacteriaceae, таких как Escherichia, Salmonella, Shigella, Citrobacter, Cronobacter, Klebsiella, Pectobacterium, то у всех них совершенно другие Ala tRNA. А именно у них Ala tRNA_GCA с Id=00011 и Ala tRNA_GCС с Id=00012. На этом основании мы и можем считать перечисленные роды семейства Enterobacteriaceae классическими представителями.
И только с родом Photorhabdus, по одной линии Ala tRNA_GCA с Id=00046, Yersinia имеет связь. Поэтому род Yersinia имеет черты от разных семейств. Такой род мы назовем переходным родом между различными семействами.
Итак, если с семейством Enterobacteriaceae род Yersinia связывает только одна конфигурация не мутировавшей Ala tRNA_GCA с Id=00046 (и то частично), то встает вопрос с каким семейством род Yersinia связан по другой конфигурации не мутировавшей Ala tRNA_GCС с Id=00043?
Оказывается наиболее прямой связью по этой линии является род Shewanella (семейство: Shewanellaceae, порядок: Alteromonadales, класс: гамма-протеобактерии). При этом объединяющая их линия Ala tRNA_GCС с Id=00043, является ключевой в эволюционном плане, так как присутствует так же еще у родов Pseudomonas и Vibrio. Все эти связи более близкие, чем следует из современной классификации, где эти роды объединены лишь на уровне класса.
В свою очередь некоторые представители рода Shewanella имеют одинаковые Ala tRNA_GCA с Id=00047 и Ala tRNA_GCС с Id=00043. Уже выяснив, что Yersinia и Shewanella имеют связь по одной линии (Id=00043), интересно с кем связан род Shewanella по другой линии (Id=00047). Оказывается, что эта линия также является достаточно ключевой в эволюционном плане. Она разветвляет происхождение далее и Ala tRNA_GCA с Id=00047 обладают так же представители родов Vibrio, Thiomicrospira, Saccharophagus.
Мы можем также проследить эти связи и далее (см. рисунок). Но уже сейчас понятно, чтобы систематизировать эти эволюционные связи, нужно несколько изменить подход к их описанию.
в более высоком разрешении
{kind=link}
Выводы
Следует еще раз подчеркнуть, что все выводы сделанные в работе, базируются на анализе только tRNA Ala, и конечно для признания результатов нужна проверка по другим видам тРНК. Но тем ни менее уже сейчас мы можем сделать некоторые выводы и описать каким образом нужно изменить подходы к систематике таксонов.
Достаточную сложность вызывает определение какой из видов более эволюционно древний, а какой более молодой. Но если исходить из гипотезы, что биологический мир развивался от простого к сложному, то у нас есть по крайней мере один неоспоримый факт. Если бактерия имеет две хромосомы, то кажется очевидным, что она эволюционно более молода, чем имеющая одну.
Поэтому в рамках нашего исследования можно с уверенностью сказать, что род Vibrio более молодой, чем Shewanella или Yersinia. И тогда, раз Vibrio и Shewanella объединены идентичными Ala tRNAGCA с Id=00047, то с большой вероятностью, род Vibrio произошел от Shewanella и так произошла одна хромосома. После чего другие представители рода Vibrio произошли от рода Colwellia и так произошла вторая хромосома. В результате объединения этих хромосом в одном организме мы и можем говорить о роде Vibrio, который по одной линии произошел от Shewanella, а по другой от Colwellia.
Таким образом, мы должны говорить не о происхождении от одного предка, а о как минимум двух, а то и больше.
С однохромосомными бактериями сложнее определить направление эволюции (кто более молодой, а кто более древний). Но основываясь на двуххромосомные виды, можно сказать, так как существует вид Vibrio с Ala tRNA_GCA с Id=00049 и Ala tRNA_GCС с Id=00043; а также существуют виды Vibrio с Ala tRNA_GCA с Id=00047, то первоночально существовали именно Ala tRNA_GCA с Id=00047 и Ala tRNA_GCС с Id=00043. А они содержались в Shewanella, и поэтому его нужно признать наиболее древним организмом, и положить в основу систематики рассмотренных здесь организмов.
Тогда можно сделать вывод, что от Shewanella по одной линии произошли Yersinia. От Yersinia в свою очередь Photorhabdus, от которого и произошло все семейство Enterobacteriaceae. Но это только по одной линии. По другой же мы уже упоминали какие роды произошли от Shewanella.
Многовидовое происхождение сильно запутывает эволюционную картину, но с этим ничего не поделаешь - такова сложность видообразования, и нам нужно лишь их наиболее точно отразить в условиях, когда не все виды известны.
upd. Ах, да. Совсем забыл, тут начали говорить - ну зачем программистам дурить голову всякой биологией. Так я собственно, и хотел заинтересовать именно программистов этой темой, так как именно они способны написать алгоритмы для биовычислений. У меня просто у самого не хватает сил провести более полный анализ. Вдруг кого-то заинтересует милости прошу пишите в личку.----------------
КАК ЖЕ ДАЛЕКО ДО ИИ. КАМЕНТЫ
КАК ЖЕ ДАЛЕКО ДО ИИ. КАМЕНТЫ2
КАК ЖЕ ДАЛЕКО ДО ИИ. КАМЕНТЫ3
--------------
отрывок
http://evolbiol.ru/latgentrans.htm
Горизонтальный перенос генов и его роль в эволюции
Долгое время эволюционная теория базировалась на представлении о том, что виды не могут обмениваться друг с другом наследственной информацией. Как только вид делится на два, и между ними возникает репродуктивная изоляция, они эволюционируют дальше сами по себе по схеме "случайные мутации + естественный отбор". На этом основывалась и Дарвиновская схема дивергенции, и столь модный на Западе кладизм - "филогенетическая систематика", и все "эволюционные деревья" и иные реконструкции исторического развития биоты. Однако расчеты показывали, что при такой изолированной эволюции отдельных видов на основе случайных мутаций и отбора жизнь просто не успела бы за сравнительно недолгий срок своего существования (4 млрд. лет) развиться от простейших форм до таких высокоорганизованных, как млекопитающие и человек.
-----------------------------
КАК ЖЕ ДАЛЕКО ДО ИИ. НЦИКЛ СТАТЕЙ 2