Проблема науки в исследовании вирусов
Mike Stone, 12 апреля 2021

Проблема для вирусологов в том, что они не могут утверждать, что то, что они представляют в качестве доказательства существования «вируса», имеет отношение к природным частицам, которые встречаются внутри живого организма...
Трудности науки в исследовании вирусов. Проблемы электронной микроскопии
Автор - Mike Stone
Огромная проблема для вирусологов заключается в том, что у них нет никакой возможности утверждать, что то, что они представляют в качестве доказательства существования «вируса», имеет какое-либо отношение к природным частицам, которые встречаются “in vivo” или внутри живого организма. Многочисленные изменяющие клетки химические вещества/антибиотики и чужеродная животная РНК, которая впоследствии смешивается с оригинальным неочищенным образцом (мазком) от больного человека, немедленно уничтожают любые претензии на очищение. Тот факт, что так много добавлено к образцу, разрушает утверждение об изоляции.
Если процесса культивирования клеток недостаточно для того, чтобы изменить образец за пределами его первоначального состояния, то разрушение естественной морфологии клеток, вызванное подготовкой образца для изображений просвечивающего электронного микроскопа, гарантирует, что это так и есть. Ниже выделены некоторые из этапов (фиксация и встраивание), через которые проходит уже измененный образец клеточной культуры для подготовки к визуализации:

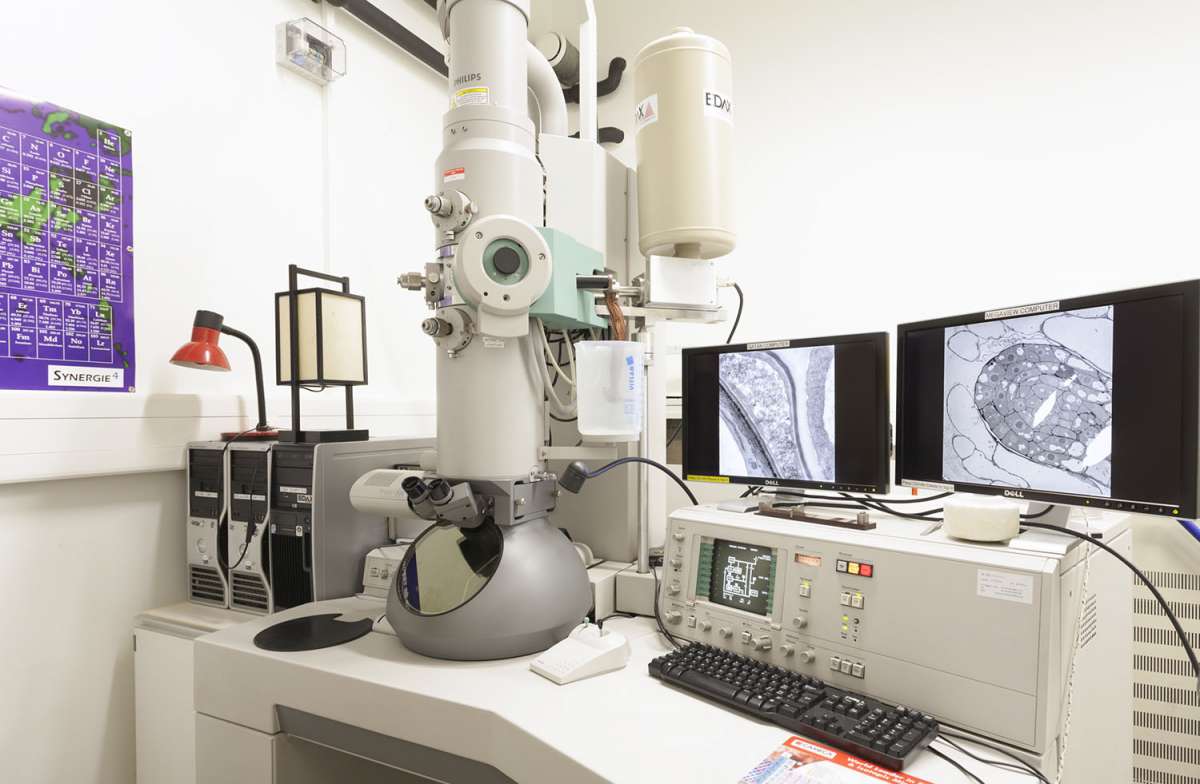
«Исследования в области вирусологии идут рука об руку с развитием методов микроскопии. Среди них большую роль сыграла электронная микроскопия (ЭМ) из-за малого размера вирусных частиц, которые, за очень редким исключением, не могут быть визуализированы обычной световой микроскопией [1,2,3,4]. До изобретения электронного микроскопа в 1931 году немецкими инженерами Эрнстом Руской и Максом Кноллем [5] ВИРУСЫ ОБНАРУЖИВАЛИСЬ ОПОСРЕДОВАННО, например, ПОСРЕДСТВОМ ЦИТОПАТИЧЕСКОГО ЭФФЕКТА, КОТОРЫЙ ОНИ ВЫЗЫВАЮТ В ИНФИЦИРОВАННЫХ КЛЕТКАХ, ИЛИ ЧЕРЕЗ КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ. Однако наличие ЭМ позволило нам визуализировать и идентифицировать многие инфекционные агенты, вызывающие заболевания или живущие “в симбиозе” с другими организмами. Таким образом, в течение 20-го века ЭМ была стандартной методикой диагностики вирусов (обзоры [6,7,8,9]).»
«Подготовка клеток к ЭМ должна следовать одной главной цели - СОХРАНИТЬ УЛЬТРАСТРУКТУРУ В СОСТОЯНИИ, МАКСИМАЛЬНО ПРИБЛИЖЕННОМ К МОМЕНТАЛЬНОМУ СНИМКУ ЖИВЫХ СОСТОЯНИЙ. Цитируя Гарета Гриффитса, первопроходца в использовании ЭМ [14]: “клеточная структура должна быть сохранена точно такой, какой она была в живом состоянии, и должна быть визуализирована на пределе разрешения электронного микроскопа”. Для достижения этой цели существует несколько методов и разработаны стандартные рецепты. Хотя они очень полезны для рутинного применения, они должны часто модифицироваться при решении конкретных биологических вопросов.»
«Стандартный метод подготовки клеток к рутинной ЭМ включает следующие этапы: ФИКСАЦИЯ, ВСТРАИВАНИЕ и СЕКЦИОНИРОВАНИЕ. Они будут подробно описаны ниже и обобщены на рис.1.»

1. ФИКСАЦИЯ КЛЕТОК
Первым и наиболее важным шагом для визуализации биологических объектов с помощью ЭМ является фиксация, поскольку ОНА ОПРЕДЕЛЯЕТ, НАСКОЛЬКО БЛИЗКО ИЗОБРАЖЕНИЕ, ВИДИМОЕ В МИКРОСКОП, НАПОМИНАЕТ СТРУКТУРУ IN VIVO. ЦЕЛЬ СОСТОИТ В ТОМ, ЧТОБЫ ИЗБЕЖАТЬ ИЛИ УМЕНЬШИТЬ ПОГРЕШНОСТИ, ВЫЗВАННЫЕ ЭКСТРАКЦИЕЙ, ДЕНАТУРАЦИЕЙ, СТЕРИЧЕСКИМИ ПОМЕХАМИ, ХИМИЧЕСКИМИ ИЗМЕНЕНИЯМИ ЭПИТОПОВ (особенно важными для иммуноцитохимического анализа) И ИЗМЕНЕНИЯМИ ОБЪЕМА И ФОРМЫ (критичными для методов 3D-анализа, описанных в этом обзоре) [14]. Фиксация клеток может осуществляться двумя различными способами: химической фиксацией (рис. 1А) или крио-иммобилизацией (т. е. физической фиксацией; рис. 1Б).
1.1 ХИМИЧЕСКАЯ ФИКСАЦИЯ
Химическая фиксация клеток обычно осуществляется буферными альдегидами. На этом этапе альдегиды создают межмолекулярную и внутримолекулярную сеть ковалентных взаимодействий (“перекрестных связей”), главным образом между аминогруппами, которые стабилизируют биологический образец. Это приводит к образованию большой трёхмерной сети необратимых поперечных связей по всей цитоплазме за время от десятых долей секунды до нескольких минут.
В то время как большинство лабораторий имеют свои собственные предпочтения, ГЛУТАРОВЫЙ АЛЬДЕГИД (ГА) ЛИБО САМ ПО СЕБЕ, ЛИБО В СОЧЕТАНИИ С ПАРАФОРМАЛЬДЕГИДОМ (ПФА) ЯВЛЯЮТСЯ НАИБОЛЕЕ ЧАСТО ИСПОЛЬЗУЕМЫМИ ПЕРВИЧНЫМИ ФИКСАТОРАМИ. Однако ГА индуцирует разветвлённую сеть перекрёстных связей, которые стерически препятствуют доступности антител к антигену. По этой причине формальдегид, который меньше маскирует антигенность, в основном используется для иммуноцитохимических исследований, таких как иммунофлуоресценция [14].
Во время всех этапов обработки очень важно, чтобы клетки не высыхали. Это особенно важно перед фиксацией, до первоначального контакта с фиксатором, когда клетки ещё живы. ХОТЯ ПРОЦЕСС ФИКСАЦИИ УБИВАЕТ КЛЕТКИ, КЛЕТКИ ДОЛЖНЫ УМИРАТЬ “ДОЛЖНЫМ ОБРАЗОМ”, ЧТОБЫ ГАРАНТИРОВАТЬ, насколько это возможно, ЧТО ВСЕ КЛЕТОЧНЫЕ КОМПОНЕНТЫ СОХРАНЯЮТСЯ ТАК ЖЕ ХОРОШО, КАК КОГДА ОНИ БЫЛИ ЖИВЫ. Для достижения этой цели можно было бы принять во внимание несколько других факторов. Следовательно, чистота альдегидов также имеет решающее значение. Поэтому рекомендуется использовать альдегиды уровня ЭМ, предоставляемые коммерческими поставщиками. Кроме того, на склеивающую способность этих альдегидов влияют время, концентрация и температура [19]. Рутинную фиксацию проводят в течение 15-30 мин при комнатной температуре с использованием буфера с физиологическим рН (6,8-7,4) и концентрацией не менее 0,1 М (моль/л) [14]. Природа буфера играет также очень важную роль при фиксации: например, наиболее широко используются какодилат натрия, фосфат натрия, ГЭПЭС (4-(2-гидроксиэтил) -1-пиперазинэтаносульфоновая кислота) и ПИПЭС (пиперазин-N, N‘-бис (2-этансульфоновая кислота)).»
“К СОЖАЛЕНИЮ, ОБЩЕГО РЕЦЕПТА ХИМИЧЕСКОЙ ФИКСАЦИИ НЕ СУЩЕСТВУЕТ. НАИЛУЧШИЕ РЕЗУЛЬТАТЫ ТРЕБУЮТ АДАПТАЦИИ К ИНДИВИДУАЛЬНЫМ УСЛОВИЯМ ЭКСПЕРИМЕНТА. Поэтому мы рекомендуем проконсультироваться со специалистом по ЭМ или свериться с литературой, чтобы помочь вам выбрать оптимальные условия (включая тип и концентрацию альдегида; тип, концентрацию и рН буфера; время и температуру фиксации), адаптированные для ваших экспериментов.
После фиксации клетки промывают предпочтительным буфером, в котором клетки могут храниться при температуре 4 °С до дальнейшей обработки. Обратите внимание, что культивируемые клетки могут быть приготовлены для ЭМ в виде монослоев или гранул (рис. 1А). Когда клетки далее обрабатываются в виде гранул, они должны быть соскоблены с чашки клеточной культуры после фиксации, если они не растут в суспензии. Альтернативно, для крио-ЭМ культивируемые клетки могут быть “трипсинизированы” перед фиксацией (рис. 1Б). СЛЕДУЕТ ИМЕТЬ В ВИДУ, ЧТО ГРАНУЛИРОВАНИЕ КЛЕТОК ИЗМЕНЯЕТ ИХ МОРФОЛОГИЮ И МОЖЕТ ПРИВЕСТИ К ОШИБКАМ (Рис. 2).»
«ОСНОВНЫМ НЕДОСТАТКОМ ХИМИЧЕСКОЙ ФИКСАЦИИ ЯВЛЯЕТСЯ ТО, ЧТО ОНА ИЗМЕНЯЕТ СТРУКТУРУ КЛЕТКИ, ОБРАЗУЯ СЕТКУ СШИТЫХ МОЛЕКУЛ. ТАКАЯ СЕТКА ПОДВЕРЖЕНА ОШИБКАМ, что является серьёзной проблемой для большинства исследований на основе ЭМ (подробное обсуждение этой темы см. в [21]). Тем не менее химическая фиксация была основой ЭМ на протяжении десятилетий, поскольку она ДОСТАТОЧНО ХОРОШО СОХРАНЯЕТ МОРФОЛОГИЮ КЛЕТОК.»
«Как уже указывал Смолл 34 года назад [24], БОЛЬШИНСТВО УЛЬТРАСТРУКТУРНЫХ ИЗМЕНЕНИЙ МОЖЕТ ПРОИСХОДИТЬ ВОВРЕМЯ ПОСТФИКСАЦИОННОЙ ОБРАБОТКИ ОБРАЗЦОВ ДЛЯ ВСТРАИВАНИЯ (описанной ниже), А НЕ ВО ВРЕМЯ ФИКСАЦИИ. Таким образом, индивидуальный протокол должен быть разработан для максимальной сохранности интересующих клеток/тканей, включая как оптимальные условия фиксации, так и условия после фиксации.»
1.2 КРИОФИКСАЦИЯ
Методы замораживания представляют собой альтернативу СКЛОННОЙ К ПОГРЕШНОСТЯМ ХИМИЧЕСКОЙ ФИКСАЦИИ (рассмотренной в работе [25]). Основной принцип заключается в том, чтобы остановить клетки путём быстрого охлаждения, процесс, который занимает несколько миллисекунд, что приводит к одновременной стабилизации всех клеточных компонентов без изменения их окружающей среды. Простейший способ заключается в погружении клеток, растущих на ЭМ-сетках, в жидкий этан или пропан путём погружения [26,27] и струйного замораживания [28,29,30,31,32] соответственно (рис. 1Б). Неотъемлемым ограничением этих подходов к быстрому охлаждению является то, что образцы могут быть покрыты только на глубину микрометров от их поверхности [33,34,35]. Это связано с плохой теплопроводностью воды: высокие скорости поверхностного охлаждения быстро распадаются внутри образца, достигая низкого значения, что вызывает кристаллизацию воды [36,37,38]. КРИСТАЛЛЫ ЛЬДА ИЗМЕНЯЮТ УЛЬТРАСТРУКТУРУ ЦИТОПЛАЗМЫ, ВЫЗЫВАЯ ФАЗОВУЮ СЕГРЕГАЦИЮ МЕЖДУ ВОДОЙ И РАСТВОРЕННЫМИ ВЕЩЕСТВАМИ [37,39]. ЕЩЁ ХУЖЕ ТО, ЧТО РОСТ КРИСТАЛЛОВ ЛЬДА МОЖЕТ ПРИВЕСТИ К ОБРАЗОВАНИЮ ДЫР В МЕМБРАНАХ И РАЗРУШЕНИЮ ОРГАНЕЛЛ [40].»
2.1 ВСТРАИВАНИЕ ХИМИЧЕСКИ НЕПОДВИЖНЫХ ЯЧЕЕК
После химической фиксации клетки должны быть дополнительно обработаны, чтобы проанализировать их с помощью ЭМ. Из-за низкой мощности рассеяния электронов биологические образцы по своей природе обладают низким контрастом [56]. Поэтому тяжёлые металлы, такие как тетроксид осмия (OsO4) и уранилацетат (UA), обладающие высоким сродством ко многим клеточным структурам, используются после фиксации в качестве контрастных агентов при рутинной ЭМ. Благодаря своей реакционной способности с ненасыщенными ацильными цепями мембранных липидов OsO4, например, способствует удержанию липидов [57,58], в дополнение к своей роли в контрастных структурах (особенно мембранах). Аналогично, Сильва и др. [59] показали, что UA играет определённую роль в защите липидов от экстракции растворителем.»
«Как было сказано ранее, на ультраструктурную сохранность клеток влияет не только способ фиксации, но и последующие стадии обработки (дегидратация, инфильтрация и полимеризация смолы). В РЕЗУЛЬТАТЕ ДЕНАТУРИРУЮЩИХ ЭФФЕКТОВ ЭТИХ СТАДИЙ ОБРАБОТКИ СТРУКТУРЫ РЕДКО СОХРАНЯЮТСЯ В ТОМ ОБЪЁМЕ, В КАКОМ ОНИ ПОЯВЛЯЮТСЯ IN VIVO».
Обратите внимание, что и фиксация, и встраивание, как известно, изменяют морфологию клеток, вносят ошибки и редко сохраняют структуры в том виде, в каком они появляются “in vivo”. Помните, что эти шаги являются дальнейшим изменением, которое было сделано в исходном образце после процесса культивирования высокотоксичных клеток.
Здесь нет ни попыток очищения, ни изоляции. Существуют миллиарды одинаковых частиц, которые могут присутствовать. Вирусологи выбирают любую сильно изменённую частицу, которая соответствует форме или идее «вируса», которую они хотят найти, и делятся ею как доказательством упомянутого «вируса».» Они не принимают во внимание, что эти частицы, скорее всего, изначально не были частью исходного образца и что они были созданы в процессе культивирования, фиксации и внедрения. Они принимают форму, функцию и патогенность, даже не доказывая этого.
Вот почему очистка/выделение неизмененного образца непосредственно от больного пациента является единственным способом гарантировать, что-то, что изображается, имеет какое-либо отношение к делу.
Источник
ПАНДЕМИЧЕСКИЙ ТЕАТР Теория заразных болезней и её криминальная история Лекция
Кровь под микроскопом после короновируса и во время него / после сауны / после ледяного душа
Более подробную и разнообразную информацию о событиях, происходящих в России, на Украине и в других странах нашей прекрасной планеты, можно получить на Интернет-Конференциях, постоянно проводящихся на сайте «Ключи познания». Все Конференции - открытые и совершенно безплатные. Приглашаем всех просыпающихся и интересующихся…
Рубрики: | Что делать? | Настоящая наука | Истинное здоровье
Постоянный адрес статьи: http://xn--b1amnebsh.ru-an.info/новости/проблема-науки-в-исследовании-вирусов-которых-может-не-быть/